The Dam-V5-LmnB1 fusion protein was verified to be co-localized with the endogenous Lamin B protein by immunofluorescence staining (Figure 1).
The successful PCR amplification of adenine-methylated DNA fragments is a key step for DamID-seq. The experimental samples should amplify a smear of 0.2 – 2 kb while the negative controls (without DpnI, without ligase or without PCR template) should result in no-or clearly less-amplification (Figure 2).
The methylated DNA fragments are in the range from 0.2 to 2 kb, while the desired insert size for an NGS library is from 200 to 300 bp. Therefore, it is essential to fragment the methyl PCR products into the suitable size range. Nonetheless, it was found to be impractical to simultaneously break larger DNA fragments down to suitable sizes and keep the majority of smaller DNA fragments intact in a single fragmentation duration. Therefore, time course experiments were performed to determine the minimal time (T0.2kb) needed to fragment 1 µg DNA to a smear centered at 200 bp (Figure 3). Then 6 time durations in equal increments were selected between 5 min and T0.2kb for the actual fragmentation. The enzymatic activity of double strand DNA Fragmentase may vary from batch to batch and may decrease over time, so it is recommended to repeat this step for a new batch of Fragmentase or after storage for a period of time.
The desired insert size is between 200 and 300 bp corresponding to DNA fragments between 300 and 400 bp (including 121 bp sequencing adaptors) on the agarose gel. Three thin slices within this range were excised from each experimental sample to narrow the size range of a library and increase the possibility of obtaining at least one qualified sequencing library (Figure 4).
An aliquot of 5 µl of each amplified DNA library was analyzed on the agarose gel to determine which library may qualify for sequencing. As shown in Figure 5A, a clear single band of the same size as the excised gel slice should be visible on the agarose gel (step 3.7.4). Next, selected libraries were examined by a Bioanalyzer (Figure 5B) to determine the exact size range and concentrations prior to sequencing. If desired, amplified DNA libraries can be directly examined by a Bioanalyzer without gel analysis. When multiple libraries are of good quality, it is recommended to sequence libraries of similar size ranges for a pair of experimental (cells expressing Dam-V5-POI) and control (cells expressing V5-Dam) samples.
The short reads generated by sequencing systems were first mapped back to the corresponding genome. Uniquely aligned reads were then passed to subsequent analyses. A pipeline to process short reads, construct a genome-NL interaction map and analyze gene-NL associations were described in detail in our previous work 10. Representative results are shown in Figure 6.
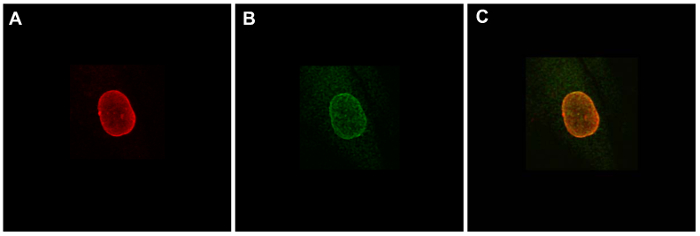
Figure 1. Validating subnuclear localizations of fusion proteins by immunofluorescence staining. IMR90 human lung fibroblast cells were transiently transfected with a plasmid expressing Dam-V5-LmnB1 and were stained by anti-Lamin B (A, red) and anti-V5 (B, green). (C) Merge of images in A and B. Please click here to view a larger version of this figure.
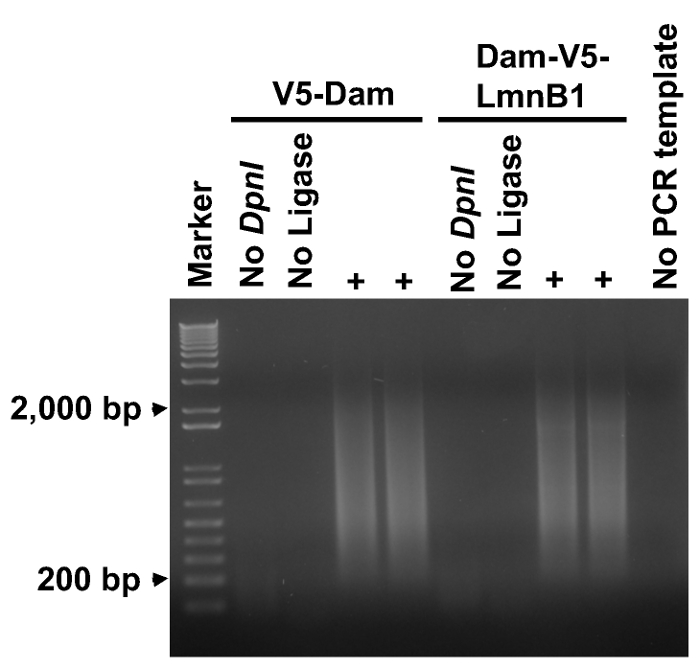
Figure 2. PCR amplifying adenine-methylated DNA fragments. An aliquot of 5 µl from each PCR reaction was analyzed on a 1% agarose gel. A smear ranging from 0.2 to 2 kb was amplified from each experimental sample, but no amplification was observed in negative controls (no DpnI in step 2.1, no Ligase in step 2.2, or no PCR template). Please click here to view a larger version of this figure.
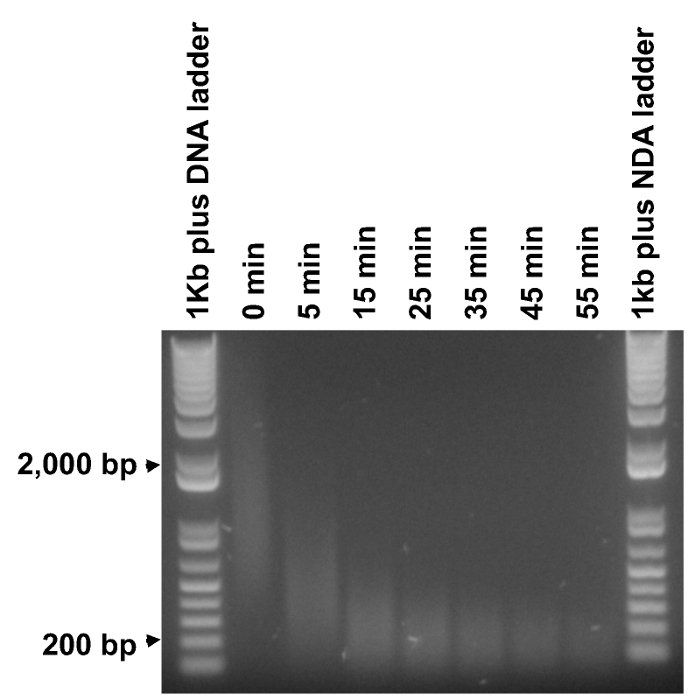
Figure 3. Determining optimal fragmentation durations. Purified methyl PCR products were subject to fragmentation for time durations from 5 to 55 min at an increment of 10 min (undigested DNA, labeled as "0 min"). 1 µg of DNA ladder and 0.5 µg of each fragmented DNA sample were analyzed on an agarose gel. The minimal time to digest the majority of the DNA smear to around 200 bp was determined to be around 35 min, therefore six evenly spaced time durations between 5 and 35 min (5 and 35 min included) were selected to perform the actual fragmentation. Please click here to view a larger version of this figure.
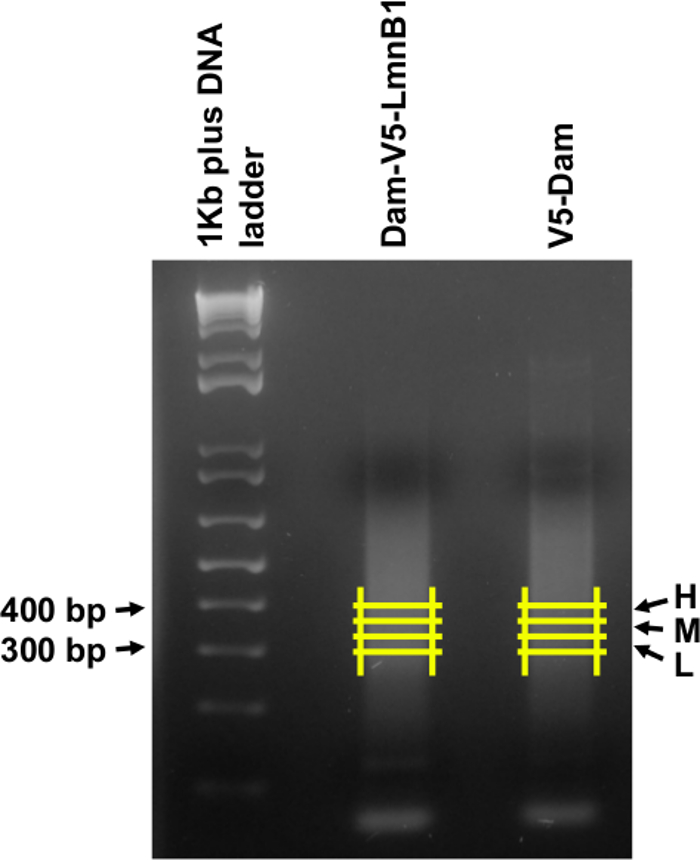
Figure 4. Size selecting the DNA libraries. DNA samples of Dam-V5-LmnB1 and V5-Dam from step 3.6 were run on a 2% agarose gel, and three equally sized gel slices (L, M and H corresponding to low, medium and high in size) were excised between 300 and 400 bp as shown by the yellow lines. Please click here to view a larger version of this figure.
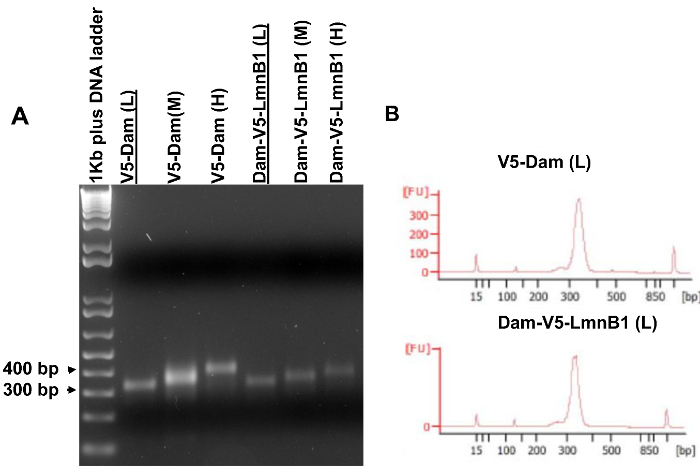
Figure 5. Amplified DNA libraries for high throughput sequencing. (A) Amplified DNA libraries analyzed on an agarose gel. PCR templates were purified from gel slices shown on Figure 4. (B) Bioanalyzer results of the V5-Dam (L) sample and the Dam-V5-LmnB1 (L) sample that were underlined in (A). These two libraries had the similar narrow size ranges and thus qualified for high throughput sequencing. Please click here to view a larger version of this figure.
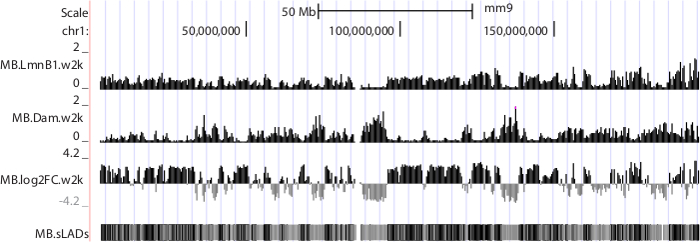
Figure 6. NGS data displayed in UCSC genome browser. Mouse chromosome 1 is shown as an example. Sequence data were produced from mouse C2C12 myoblasts 10. Tracks "MB.LmnB1.w2k" and "MB.Dam.w2k", corresponding to data from cells expressing Dam-V5-LmnB1 and V5-Dam respectively, plot normalized read densities (reads per kilobase per million uniquely mapped reads, or RPKM) in non-overlapping consecutive 2 kb windows along the chromosome. Track "MB.log2FC.w2k" plots genome-NL associations, i.e. log2 RPKM ratios of LmnB1 over Dam, at 2 kb resolution. Track "MB.sLADs" paints sequencing-based Lamina Associated Domains (sLADs, i.e. genomic regions that have higher read densities of LmnB1 over Dam with statistical significance) in black, non-sLADs in gray and undetermined regions in white. Please click here to download this file.
