Одним из самых захватывающих открытий за последние два десятилетия в биологии является распространяющаяся роль видов sRNA в регулировании различных функций генома1. В частности, miRNAs представляют собой важный класс от 20 до 24-nt sRNAs в эукариоты, и в основном функции на посттранскрипционном уровне в качестве видных регуляторов генов на протяжении всей стадии развития жизненного цикла, а также в стимулировочных и стрессовых реакций2,3. В растениях, miRNAs возникают из первичных стенограмм ы называется pri-miRNAs, которые, как правило, транскрибируется РНК полимеразы II как отдельные блоки транскрипции4,5. Обработанные эволюционно сохраненным клеточным оборудованием (Drosha RNase III у животных, DICER-как в растениях), при-миРНК иссечены в непосредственные прекурсоры miRNA, предварительно миРНК, которые содержат последовательности, образующие внутримолекулярные структуры стволовой петли6,7. Pre-miRNAs затем обрабатываются в двухцепочечные промежуточные, а именно miRNA дуплексы, состоящие из функциональной нити, зрелые miRNA, и реже функциональный партнер, miRNA2,8. После загрузки в РНК-индуцированной глушитель комплекса (RISC), зрелые miRNAs может распознавать их мРНК цели на основе последовательности взаимодополняемости, в результате чего негативная регулятивная функция2,8. miRNAs может либо дестабилизировать свои целевые стенограммы или предотвратить целевой перевод, но в первом случае доминируют в растениях8,9.
С случайного открытия первого miRNA в nematode Caenorhabditis elegans10,11, много исследований было совершено для идентификации miRNA и его функциональный анализ, особенно после наличия метода NGS. Широкое применение метода NGS значительно способствовало использованию вычислительных инструментов, которые были разработаны для захвата уникальной особенностью miRNAs, таких как структура стволовых петли прекурсоров и их преференциальное накопление последовательности читает на зрелых miRNA и miRNA. В результате исследователи добились значительных успехов в выявлении miRNAs у различных видов. Основываясь на ранее описанной вероятностной модели12, мы разработали miRDeep-P13, который был первым вычислительным инструментом для обнаружения растительных миРНК из данных NGS. miRDeep-P был специально направлен на завоевание проблем декодирования завода miRNAs с более переменной длиной прекурсоров и большими парализующими семьями13,14,15. После его выпуска, эта программа была загружена тысячи раз и используется для аннотации miRNA транскриптомы в более чем 40 видов растений16. Propelled NGS-основанных инструментов, таких как miRDeep-P, наблюдается резкое увеличение числа зарегистрированных miRNAs в общественном репозитории miRNA miRBase17, где более 38000 miRNA пунктов в настоящее время размещается (выпуск 22.1) по сравнению только с 500 miRNA пунктов (выпуск 2.0) в 200818.
Тем не менее, две новые проблемы возникли из аннотации miRNA завода. Во-первых, высокие коэффициенты ложных срабатываний оказали значительное влияние на качество аннотации миннакирастений 16,19 по следующим причинам: 1) поток эндогенных короткой интерферинговых РНК (siRNAs) из библиотек NGS sRNA были ошибочно аннотированы как миРНК из-за отсутствия строгих критериев аннотации миРНК; 2) для видов без информации miRNA, ложные срабатывания предсказанные на основе данных NGS трудно устранить. Используя miRBase в качестве примера, Taylor et al.20 обнаружили, что одна треть записей растительной miRNA в общественном хранилище21 (выпуск 21) не имеет убедительных подтверждающих доказательств, и даже три четверти семейсемей сярей miRNA были сомнительными. Во-вторых, он становится чрезвычайно трудоемким процессом для прогнозирования растительных миРНК с большими и сложными геномами16. Чтобы преодолеть эти проблемы, мы обновили miRDeep-P, добавив новую стратегию фильтрации, перепланировку алгоритма скоринга и интеграцию новых критериев для аннотации miRNA завода, и выпустили новую версию miRDP2. Кроме того, мы протестировали miRDP2 с использованием наборов данных NGS sRNA с постепенным увеличением размеров генома: арабидопсис, рис, помидоры, кукуруза и пшеница. По сравнению с другими пятью широко используемыми инструментами и его старой версией, miRDP2 разогнал эти данные sRNA и проанализировал транскриптомы miRNA быстрее с улучшенной точностью и чувствительностью.
Содержимое пакета miRDP2
Пакет miRDP2 состоит из шести задокументированных скриптов Perl, которые должны быть запущены последовательно подготовленным скриптом Bash. Из шести скриптов три(convert_bowtie_to_blast.pl, filter_alignments.pl,и excise_candidate.pl)унаследованы от miRDeep-P. Другие скрипты изменены из исходной версии. Функции шести скриптов описаны следующим образом:
preprocess_reads.pl фильтры ввода читает, в том числе читает, которые являются слишком длинными или слишком короткими (Злт;19 nt или йgt;25 nt), и читает коррелирует с Rfam ncRNA последовательностей, а также читает с RPM (читает на миллион) менее 5. Затем скрипт извлекает считываемые, коррелированные с известными зрелыми последовательностями miRNA. Файлы ввода являются оригинальными считываемыми в формате FASTA/FASTи и выходом bowtie2 считываний на miRNA и ncRNA последовательностей.
Формула расчета RPM:
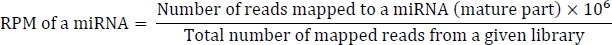
convert_bowtie_to_blast.pl изменяет формат bowtie в BLAST-разбора формата. BLAST-parsed формат представляет собой пользовательский табулярный разделенный формат, полученный от стандартного формата NCBI BLASToutput.
filter_alignments.pl фильтрует выравнивания глубокого секвенирования считывает на геном. Он фильтрует частичные выравнивания, а также многовыровные считывавшие ся (определенное пользователем частотное отсечение). Основным входом является файл в формате BLAST-parsed.
excise_candidate.pl вырезает потенциальные последовательности прекурсоров из референтной последовательности, используя выровненные чтения в качестве руководящих принципов. Основным входом является файл в формате BLAST-parsed и файл FASTA. Выход — это все потенциальные последовательности прекурсоров в формате FASTA.
mod-miRDP.pl требуется два файла ввода, файл подписи и файл структуры, который изменяется из основного алгоритма miRDeep-P путем изменения системы скоринга с конкретными параметрами завода. Файлы ввода представляют сяртово-кронштейнный файл структуры прекурсоров и считывают файл подписи распределения.
mod-rm_redundant_meet_plant.pl нуждается в трех файлах ввода: chromosome_length, прекурсорах и original_prediction, генерируемых mod-miRDP.pl. Он генерирует два выходных файла, неизлишний прогнозируемый файл и прогнозируемый файл, отфильтрованный по недавно обновленным критериям miRNA завода. Подробная информация о формате вывода файла описана в разделе 1.4.
