Eine der spannendsten Entdeckungen der letzten zwei Jahrzehnte in der Biologie ist die sich ausbreitende Rolle von sRNA-Arten bei der Regulierung verschiedener Funktionen des Genoms1. Insbesondere stellen miRNAs eine wichtige Klasse von 20- bis 24-nt sRNAs in Eukaryoten dar und fungieren hauptsächlich auf posttranskriptionärer Ebene als prominente Genregulatoren während der gesamten Lebenszyklusentwicklungsphase sowie in Stimulus- und Stressreaktionen2,3. Bei Pflanzen entstehen miRNAs aus primären Transkripten, sogenannten pri-miRNAs, die in der Regel durch RNA-Polymerase II als individuelle Transkriptionseinheiten4,5transkribiert werden. Verarbeitet durch evolutionär konservierte zelluläre Maschinen (Drosha RNase III bei Tieren, DICER-like in Pflanzen) werden pri-miRNAs in die unmittelbaren miRNA-Vorstufen, pre-miRNAs, eingeschnitten, die Sequenzen enthalten, die intramolekulare Stammschleifenstrukturenbilden 6,7. Pre-miRNAs werden dann zu doppelsträngigen Zwischenprodukten verarbeitet, nämlich miRNA-Duplexen, bestehend aus dem funktionellen Strang, der reifen miRNA und dem seltener funktionellen Partner miRNA*2,8. Nach dem Einladen in den RNA-induzierten Silencing-Komplex (RISC) konnten die reifen miRNAs ihre mRNA-Ziele anhand der Sequenzkompleärität erkennen, was zu einer negativen Regulatorischen Funktion2,8führte. miRNAs könnten entweder ihre Zieltranskripte destabilisieren oder Zielübersetzungen verhindern, aber die frühere Art und Weise wird in den Anlagen8,9dominiert.
Seit der zufälligen Entdeckung der ersten miRNA in der Nematode Caenorhabditis elegans10,11wurde viel Forschung zur miRNA-Identifikation und ihrer funktionellen Analyse, insbesondere nach der Verfügbarkeit der NGS-Methode, durchgeführt. Die breite Anwendung der NGS-Methode hat die Nutzung von Rechenwerkzeugen, die entwickelt wurden, um die einzigartige Eigenschaft von miRNAs zu erfassen, wie die Stammschleifenstruktur von Vorläufern und ihre bevorzugte Akkumulation von Sequenzlesungen auf ausgereifter miRNA und miRNA* stark gefördert. Als Ergebnis haben die Forscher bemerkenswerte Erfolge bei der Identifizierung von miRNAs in verschiedenen Arten erzielt. Basierend auf einem zuvor beschriebenen Wahrscheinlichkeitsmodell12entwickelten wir miRDeep-P13, das erste Rechenwerkzeug zur Erkennung von pflanzlichen miRNAs aus NGS-Daten. miRDeep-P zielte speziell darauf ab, die Herausforderungen der Dekodierung von pflanzlichen miRNAs mit variablerer Vorläuferlänge und großen paralogusfarbenen Familien13,14,15zu meistern. Nach seiner Veröffentlichung wurde dieses Programm tausende Male heruntergeladen und verwendet, um miRNA-Transkriptome bei mehr als 40 Pflanzenarten16zu kommentieren. Angetrieben von NGS-basierten Tools wie miRDeep-P, hat die Anzahl der registrierten miRNAs im öffentlichen miRNA-Repository miRBase17, wo derzeit über 38.000 miRNA-Elemente gehostet werden (Release 22.1), im Vergleich zu nur 500 MIRNA-Elementen (Release 2.0) im Jahr 200818erhöht.
Allerdings sind zwei neue Herausforderungen aus der pflanzlichen miRNA-Anmerkung entstanden. Erstens haben hohe Verhältnisse von Falsch-Positiven die Qualität der pflanzlichen miRNA-Anmerkungen16,19 aus folgenden Gründen stark beeinflusst: 1) eine Flut von endogenen kurz störenden RNAs (siRNAs) aus NGS sRNA-Bibliotheken wurde fälschlicherweise als miRNAs bezeichnet, da keine strengen miRNA-Anmerkungskriterien vorliegen; 2) Für Arten ohne a priori miRNA-Informationen sind falsch positive Vorhersagen, die auf NGS-Daten basieren, schwer zu eliminieren. Am Beispiel von miRBase fanden Taylor et al.20 ein Drittel der pflanzlichen miRNA-Einträge im öffentlichen Endlager21 (Release 21) ohne überzeugende Belege und sogar drei Viertel der pflanzlichen miRNA-Familien waren fragwürdig. Zweitens wird es zu einem extrem zeitaufwändigen Prozess zur Vorhersage von pflanzlichen miRNAs mit großen und komplexen Genomen16. Um diese Herausforderungen zu meistern, haben wir miRDeep-P aktualisiert, indem wir eine neue Filterstrategie hinzugefügt, den Bewertungsalgorithmus überarbeitet und neue Kriterien für die pflanzliche miRNA-Anmerkung integriert und die neue Version miRDP2 veröffentlicht haben. Darüber hinaus haben wir miRDP2 mit NGS sRNA-Datensätzen mit allmählich steigenden Genomgrößen getestet: Arabidopsis, Reis, Tomaten, Mais und Weizen. Im Vergleich zu anderen fünf weit verbreiteten Werkzeugen und seiner alten Version analysierte miRDP2 diese sRNA-Daten und analysierte miRNA-Transkriptome schneller mit verbesserter Genauigkeit und Empfindlichkeit.
Inhalt des miRDP2-Pakets
Das miRDP2-Paket besteht aus sechs dokumentierten Perl-Skripten, die sequenziell vom vorbereiteten Bash-Skript ausgeführt werden sollen. Von den sechs Skripten werden drei (convert_bowtie_to_blast.pl, filter_alignments.plund excise_candidate.pl) von miRDeep-P geerbt. Die anderen Skripts werden ab der Originalversion geändert. Die Funktionen der sechs Skripte werden im Folgenden beschrieben:
preprocess_reads.pl filtert Eingabelesevorgänge, einschließlich Lesevorgänge, die zu lang oder zu kurz sind (25 nt), und liest korreliert mit Rfam ncRNA-Sequenzen sowie Lesevorgänge mit RPM (Reads Per Million) kleiner als 5. Das Skript ruft dann Lesevorgänge ab, die mit bekannten miRNA-Reifensequenzen korreliert sind. Die Eingabedateien sind Originallesevorgänge im FASTA/FASTQ-Format und bowtie2-Ausgabe von Lesekarten, die zu miRNA- und ncRNA-Sequenzen mapping.
Die Formel für die Berechnung von RPM lautet wie folgt:
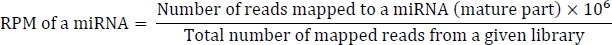
convert_bowtie_to_blast.pl ändert das Bowtie-Format in ein BLAST-parsed-Format. BLAST-parsed Format ist ein benutzerdefiniertes tabellarisches separates Format, das vom Standard-NCBI BLASToutput-Format abgeleitet wird.
filter_alignments.pl filtert die Ausrichtungen von Tiefensequenzierungslesungen zu einem Genom. Es filtert Partielle Ausrichtungen sowie mehrstimmige Lesevorgänge (benutzerspezifische Frequenzabschaltung). Die Basiseingabe ist eine Datei im BLAST-parsed Format.
excise_candidate.pl schneidet potenzielle Vorläufersequenzen aus einer Referenzsequenz unter Verwendung ausgerichteter Lesevorgänge als Richtlinien aus. Die Basiseingabe ist eine Datei im BLAST-parsed-Format und eine FASTA-Datei. Die Ausgabe sind alle potenziellen Vorläufersequenzen im FASTA-Format.
mod-miRDP.pl benötigt zwei Eingabedateien, Signaturdatei und Strukturdatei, die aus dem kernigen miRDeep-P-Algorithmus geändert wird, indem das Bewertungssystem mit pflanzenspezifischen Parametern geändert wird. Die Eingabedateien sind dot-bracket Precursor-Strukturdatei und liest VerteilungSignatur-Datei.
mod-rm_redundant_meet_plant.pl benötigt drei Eingabedateien: chromosome_length, Vorläufer und original_prediction, die von mod-miRDP.pl generiert werden. Es generiert zwei Ausgabedateien, nicht redundante vorhergesagte Datei und vorhergesagte Datei gefiltert durch neu aktualisierte plant miRNA Kriterien. Details zum Format der Ausgabedatei werden in Abschnitt 1.4 beschrieben.
