Uma das descobertas mais emocionantes nas últimas duas décadas em biologia é o papel proliferante das espécies de sRNA na regulação de diversas funções do genoma1. Em particular, os miRNAs constituem uma classe importante de 20 a 24-nt sRNAs em eucariontes, e principalmente funcionam em nível pós-transcricional como reguladores genéticos proeminentes ao longo dos estágios de desenvolvimento do ciclo de vida, bem como em respostas de estímulo e estresse2,3. Nas plantas, miRNAs surgem de transcrições primárias chamadas pri-miRNAs, que geralmente são transcritas pela RNA polymerase II como unidades de transcrição individuais4,5. Processado sigativamente conservado máquinas celulares (Drosha RNase III em animais, DICER-like em plantas), pri-miRNAs são extirbolsados para os precursores miRNA imediatos, pré-miRNAs, que contêm seqüências que formam estruturas intra-moleculares de laço-tronco6,7. Pré-miRNAs são então processados em intermediários duplos, ou seja, duplex miRNA, consistindo da vertente funcional, miRNA maduro, e o parceiro menos freqüentemente funcional, miRNA *2,8. Depois de carregados no complexo de silenciamento induzido pelo RNA (RISC), os miRNAs maduros puderam reconhecer seus alvos mRNA com base na complementaridade da sequência, resultando em uma função regulatória negativa2,8. miRNAs poderia desestabilizar suas transcrições alvo ou impedir a tradução alvo, mas a maneira anterior é dominada em plantas8,9.
Desde a descoberta fortuita do primeiro miRNA no nematóide Caenorhabditis elegans10,11, muita pesquisa tem sido comprometida com a identificação miRNA e sua análise funcional, especialmente após a disponibilidade do método NGS. A ampla aplicação do método NGS promoveu muito a utilização de ferramentas computacionais que foram projetadas para capturar a característica única dos miRNAs, como a estrutura de precursores de stem-loop e seu acúmulo preferencial de leituras de sequências em miRNA e miRNA maduros*. Como resultado, os pesquisadores alcançaram um sucesso notável na identificação de miRNAs em diversas espécies. Com base em um modelo de probabilidade descrito anteriormente12,desenvolvemos o miRDeep-P13,que foi a primeira ferramenta computacional para a descoberta de miRNAs de plantas a partir de dados NGS. o miRDeep-P visava especificamente conquistar os desafios da decodificação de miRNAs de plantas com maior comprimento de precursor e grandes famílias paralogous13,14,15. Após seu lançamento, este programa foi baixado milhares de vezes e usado para anotar transcriptomas miRNA em mais de 40 espécies de plantas16. Impulsionado por ferramentas baseadas em NGS como miRDeep-P, tem havido um aumento dramático no número de miRNAs registrados no miRBase repositório miRBASE17público , onde mais de 38.000 itens miRNA estão atualmente hospedados (lançamento 22,1) em comparação com apenas ~ 500 itens miRNA (lançamento 2.0) em 200818.
No entanto, dois novos desafios surgiram a partir da anotação de miRNA de plantas. Em primeiro lugar, as elevadas proporções de falsos positivos têm impactado fortemente a qualidade das anotações de miRNA de plantas16,19 pelas seguintes razões: 1) um dilúvio de RNAs endógenos de interferência curta (siRNAs) de bibliotecas NGS sRNA foram erroneamente anotados como miRNAs devido à falta de uma rigorosa critérios de annotação miRNA; 2) para espécies sem informações priori miRNA, os falsos positivos previstos com base em dados ngs são difíceis de eliminar. Usando miRBase como exemplo, Taylor et al.20 encontraram um terço das entradas de miRNA de plantas no repositório público21 (lançamento 21) não tinham provas convincentes de apoio e até três quartos das famílias de miRNA de plantas eram questionáveis. Em segundo lugar, torna-se um processo extremamente demorado para prever miRNAs vegetais com genomas grandes e complexos16. Para superar esses desafios, atualizamos o miRDeep-P adicionando uma nova estratégia de filtragem, revisando o algoritmo de pontuação e integrando novos critérios para a anotação de miRNA de plantas e lançou a nova versão miRDP2. Além disso, testamos miRDP2 usando conjuntos de dados NGS sRNA com tamanhos genômicos aumentando gradualmente: Arabidopsis, arroz, tomate, milho e trigo. Em comparação com outras cinco ferramentas amplamente utilizadas e sua versão antiga, o miRDP2 analisou esses dados de sRNA e analisou transcriomas miRNA mais rapidamente com maior precisão e sensibilidade.
Conteúdo do pacote miRDP2
O pacote miRDP2 consiste em seis scripts Perl documentados que devem ser executados sequencialmente pelo script bash preparado. Dos seis scripts, três(convert_bowtie_to_blast.pl, filter_alignments.ple excise_candidate.pl)são herdados do miRDeep-P. Os outros scripts são modificados a partir da versão original. As funções dos seis scripts são descritas no seguinte:
preprocess_reads.pl filtros de entrada lê, incluindo leituras que são muito longas ou muito curtas (25 nt), e lê correlacionado com seqüências de Rfam ncRNA, bem como lê com RPM (Lê Per Million) menos de 5. O script, em seguida, recupera lê correlacionado s sequências conhecidas miRNA maduro. Os arquivos de entrada são leituras originais em formato FASTA/FASTQ e saída bowtie2 de leituras de mapeamento para sequências de miRNA e ncRNA.
A fórmula para calcular o RPM é como a seguinte:
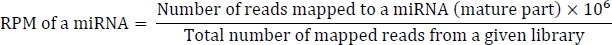
convert_bowtie_to_blast.pl muda o formato de gravata borboleta em formato BLAST-parsed. O formato blast-analisado é um formato separado tabular personalizado derivado do formato padrão NCBI BLASToutput.
filter_alignments.pl filtra os alinhamentos de seqüenciamento profundo lê a um genoma. Ele filtra alinhamentos parciais, bem como leituras multi-alinhadas (corte de frequência especificado pelo usuário). A entrada básica é um arquivo em formato BLAST-analisados.
excise_candidate.pl corta sequências precursoras potenciais de uma sequência de referência usando leituras alinhadas como diretrizes. A entrada básica é um arquivo em formato BLAST-analisados e um arquivo FASTA. A saída é todas as sequências precursoras potenciais no formato FASTA.
mod-miRDP.pl precisa de dois arquivos de entrada, arquivo de assinatura e arquivo de estrutura, que é modificado a partir do algoritmo miRDeep-P principal, alterando o sistema de pontuação com parâmetros específicos da planta. Os arquivos de entrada são arquivo de estrutura precursora do suporte de pontos e lê em arquivo de assinatura de distribuição.
mod-rm_redundant_meet_plant.pl precisa de três arquivos de entrada: chromosome_length, precursores e original_prediction gerados por mod-miRDP.pl. Ele gera dois arquivos de saída, arquivo previsto não redundante e arquivo previsto filtrado por critérios de miRNA de plantas recém-atualizados. Detalhes sobre o formato do arquivo de saída são descritos na seção 1.4.
