Uno de los descubrimientos más emocionantes de las últimas dos décadas en biología es el papel proliferante de las especies de ARNS en la regulación de diversas funciones del genoma1. En particular, los miRNAs constituyen una clase importante de ARN de 20 a 24 nt en eucariotas, y funcionan principalmente a nivel post-transcripcional como reguladores genéticos prominentes a lo largo de las etapas de desarrollo del ciclo de vida, así como en respuestas de estímulo y estrés2,3. En las plantas, los miRNAs surgen de transcripciones primarias llamadas pri-miRNAs, que generalmente son transcritas por ARN polimerasa II como unidades de transcripción individuales4,5. Procesados por maquinaria celular evolutivamente conservada (Drosha RNase III en animales, como DICER en plantas), los pri-miRNAs se extirpan en los precursores inmediatos del miRNA, pre-miRNAs, que contienen secuencias que forman estructuras de bucle de tallo intramolecular6,7. Los pre-miRNA se procesan en intermedios de doble cadena, a saber, dúplex de miRNA, que consisten en la hebra funcional, miRNA maduro y el socio menos frecuentemente funcional, miRNA*2,8. Después de cargarlos en el complejo de silenciamiento inducido por ARN (RISC), los miRNAs maduros podrían reconocer sus objetivos de ARNm en función de la complementariedad de la secuencia, lo que dio lugar a una función reguladora negativa2,8. los miRNAs podrían desestabilizar sus transcripciones objetivo o impedir la traducción de objetivos, pero la forma anterior está dominada en las plantas8,9.
Desde el descubrimiento fortuito del primer miRNA en el nematodo Caenorhabditis elegans10,11, se ha investigado mucho con la identificación de miRNA y su análisis funcional, especialmente después de la disponibilidad del método NGS. La amplia aplicación del método NGS ha promovido en gran medida la utilización de herramientas computacionales que fueron diseñadas para capturar la característica única de los miRNAs, como la estructura de bucle de vástago de precursores y su acumulación preferencial de lecturas de secuencia en miRNA maduro y miRNA*. Como resultado, los investigadores han logrado un éxito notable en la identificación de miRNAs en diversas especies. Basándonos en un modelo de probabilidad12descrito anteriormente, desarrollamos miRDeep-P13, que fue la primera herramienta computacional para descubrir miRNAs de plantas a partir de datos NGS. miRDeep-P estaba específicamente dirigido a la conquista de los desafíos de la decodificación de miRNAs vegetales con longitud precursora más variable y grandes familias paralocitas13,14,15. Después de su lanzamiento, este programa ha sido descargado miles de veces y utilizado para anotar transcriptomes de miRNA en más de 40 especies de plantas16. Propulsado por herramientas basadas en NGS como miRDeep-P, se ha producido un aumento espectacular en el número de miRNAs registrados en el repositorio público miRNAmiRBase 17,donde actualmente se alojan más de 38.000 elementos de miRNA (versión 22.1) en comparación con sólo 500 elementos de miRNA (versión 2.0) en 200818.
Sin embargo, han surgido dos nuevos desafíos a partir de la anotación del miRNA vegetal. En primer lugar, las altas proporciones de falsos positivos han afectado en gran medida la calidad de las anotaciones de miRNA vegetal16,19 por las siguientes razones: 1) un diluvio de ARN (ARN) de interferencia corta endógena (SIRNAs) de las bibliotecas de ARNS de NGS fueron erróneamente anotados como miRNAs debido a la falta de un criterio de anotación de miRNA estricto; 2) para las especies sin información de miRNA a priori, los falsos positivos previstos sobre la base de los datos de NGS son difíciles de eliminar. Utilizando miRBase como ejemplo, Taylor yotros 20 encontraron que un tercio de las entradas de miRNA de plantas en el repositorio público21 (versión 21) carecían de pruebas de apoyo convincentes e incluso tres cuartas partes de las familias de miRNA de plantas eran cuestionables. En segundo lugar, se convierte en un proceso extremadamente lento para predecir miRNAs vegetales con genomas grandes y complejos16. Para superar estos desafíos, actualizamos miRDeep-P agregando una nueva estrategia de filtrado, revisando el algoritmo de puntuación e integrando nuevos criterios para la anotación de miRNA de planta, y lanzamos la nueva versión miRDP2. Además, probamos miRDP2 utilizando conjuntos de datos de ARN NGS con tamaños de genoma sin aumento gradual: Arabidopsis, arroz, tomate, maíz y trigo. En comparación con otras cinco herramientas ampliamente utilizadas y su versión antigua, miRDP2 analizó estos datos de ARNS y analizó los transcriptomas de miRNA más rápido con una precisión y sensibilidad mejoradas.
Contenido del paquete miRDP2
El paquete miRDP2 consta de seis scripts Perl documentados que el script bash preparado debe ejecutar secuencialmente. De los seis scripts, tres (convert_bowtie_to_blast.pl, filter_alignments.ply excise_candidate.pl) se heredan de miRDeep-P. Los otros scripts se modifican de la versión original. Las funciones de los seis scripts se describen a continuación:
preprocess_reads.pl filtra las lecturas de entrada, incluidas las lecturas demasiado largas o demasiado cortas (25 nt), y lee correlacionadas con secuencias de Rfam ncRNA, así como lecturas con RPM (lecturas por millón) inferiores a 5. A continuación, el script recupera las lecturas correlacionadas con secuencias maduras de miRNA conocidas. Los archivos de entrada son lecturas originales en formato FASTA/FASTQ y salida bowtie2 de asignaciones de lecturas a secuencias miRNA y ncRNA.
La fórmula para calcular RPM es la siguiente:
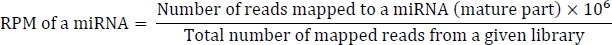
convert_bowtie_to_blast.pl cambia el formato bowtie al formato BLAST-parsed. El formato analizado por BLAST es un formato separado por tabular personalizado derivado del formato NCBI BLASToutput estándar.
filter_alignments.pl filtra las alineaciones de las lecturas de secuenciación profunda a un genoma. Filtra las alineaciones parciales, así como las lecturas multialineadas (corte de frecuencia especificado por el usuario). La entrada básica es un archivo en formato BLAST-parsed.
excise_candidate.pl elimina las posibles secuencias precursoras de una secuencia de referencia utilizando lecturas alineadas como directrices. La entrada básica es un archivo en formato BLAST-parsed y un archivo FASTA. La salida es todas las secuencias precursoras potenciales en formato FASTA.
mod-miRDP.pl necesita dos archivos de entrada, archivo de firma y archivo de estructura, que se modifica desde el algoritmo de miRDeep-P principal cambiando el sistema de puntuación con parámetros específicos de la planta. Los archivos de entrada son un archivo de estructura precursor de corchete de punto y lee el archivo de firma de distribución.
mod-rm_redundant_meet_plant.pl necesita tres archivos de entrada: chromosome_length, precursores y original_prediction generados por mod-miRDP.pl. Genera dos archivos de salida, el archivo predicho no redundante y el archivo predicho filtrado por los criterios de miRNA de planta recién actualizados. Los detalles sobre el formato del archivo de salida se describen en la sección 1.4.
