The microfabrication process consists of substrate cleaning, surface functionalization, CYTOP coating, photolithography, dry etching, photoresist stripping, and final cleaning. Importantly, the presented protocol allowed complete removal of the hydrophobic CYTOP polymer inside the microchambers Figure 3A), producing a highly parallel hydrophilic-in-hydrophobic structure on a standard cover glass substrate. With the aid of the oil sealing protocol, the uniform dimension of the resulting droplets was verified by encapsulating fluorescent solution in the microchambers (Figure 3B). The fluorescence intensity extracted using the developed software is a good indicator of the droplet size. The CV of the fluorescence intensity, 3%, reflected the narrow distribution of the droplet size over the entire array (Figure 3C). The reconstructed 3D image from confocal microscopy also showed the consistent droplet volume over time (Figure 3D, Supplementary Movie 1)44. In comparison, a widely used FC-40 oil generated the droplets exhibiting a severalfold difference in the fluorescence intensity45. The formed droplets in FemDA were stable at RT for at least 24 hours (Figure 3E). The high quality of the droplets eventually formed the basis of quantitative measurement.
High-throughput data necessitate high-throughput data-analyzing tools46,47. The developed plugin greatly simplified and speeded up the image data analysis. Based on the theory of digital image processing48, the defocused BF image can be binarized and used to provide the coordinate information of every droplet after background correction and noise reduction (Figure 4A-C). This idea made the precise localization of dark droplets possible.
The fluorescent protein mNeonGreen was synthesized in FemDA. Template DNA with a concentration of 0.05 molecules per droplet was randomly distributed into each droplet to initiate the protein synthesis with coupled cell-free transcription and translation reactions. Because the average number of DNA molecules per droplet was smaller than 1, some droplets contained zero template DNA, while others contained one or more DNA molecules. After 6 hours of incubation at RT, the end-point image was captured using the microscope. The stack image data was analyzed by the developed software with the aid of the concurrent defocused BF image (Figure 4A-E). The fluorescence intensity of each droplet is a measure of the protein yield in the corresponding droplet. The histogram of the fluorescence intensities showed a discrete distribution and was well fitted by a sum of Gaussian distributions of equal peak-to-peak intervals (Figure 4F), which strongly suggested an occupancy of different numbers of DNA molecules per droplet. Similar to the repeated coin-flipping game, the number of independent random events that occur is mathematically described by the Poisson distribution. The probability of occurrence of droplets containing different numbers (up to 3 in this example) of DNA molecules was a perfect fit to a Poisson distribution (P = e-λ • λk / k!, where λ is the expected average number of DNA molecules per droplet and k is the actual number of DNA molecules in a droplet) with an average of 0.05 DNA molecules per droplet (Figure 4G)31, as expected for a random distribution of DNA molecules. Because the loading concentration of the template DNA solution was the same as the final fitted λ (i.e., 0.05), the CFPS reaction efficiency in our FemDA was 100% (or near 100%). In other words, a single DNA molecule is enough to trigger the CFPS reaction in the femtoliter droplet efficiently.
The CFPS reaction of alkaline phosphatase was recorded every 5 minutes. The developed software also supports analyzing the time-course data (Figure 5). The coupled fluorogenic reaction showed a similar discrete distribution of the fluorescence intensity of the droplets at earlier time-points. The histogram results also verified an occupancy of different numbers of DNA molecules in the droplet. The fluorescence intensity eventually converged to a value along with the gradual depletion of the fluorogenic substrate DiFMUP.
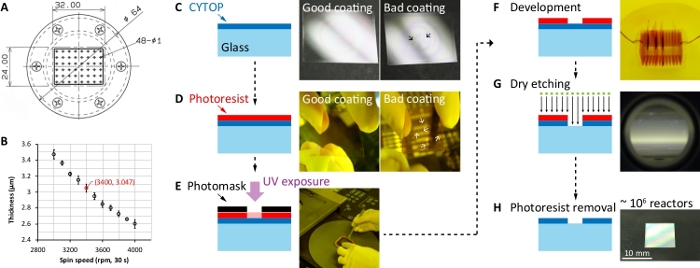
Figure 1: Microfabrication process of the ultrahigh-density microchamber array substrate. (A) Technical drawing of the customized vacuum chuck. Unit: mm. (B) CYTOP film thickness vs. spin speed data. (C) Spin-coating of the perfluoropolymer CYTOP on a silanized glass substrate. The photographs showed a good example of homogenous coating and a bad example of inhomogeneous coating, respectively. The black arrow indicated the specific position of the inhomogeneous CYTOP film. (D) Spin-coating of photoresist on the CYTOP-coated substrate. After the spin-coating, the photoresist near the substrate edge must be removed using an ethanol-soaked clean wiper (middle photograph). The photographs showed a good example of homogenous coating and a bad example of inhomogeneous coating, respectively. The white arrow indicated the specific position of the inhomogeneous photoresist film. (E) Exposure of the coated photoresist using a mask aligner. (F) Development of the exposed photoresist in a developer. The exposed part of the photoresist is soluble in the developer solution. (G) Reactive-ion etching of CYTOP. The uncovered CYTOP was removed by O2 plasma. (H) Removal of the photoresist mask. The photoresist was removed by acetone. The substrate was cleaned using 2-propanol and H2O. Please click here to view a larger version of this figure.

Figure 2: Preparation and use of the microchamber array device. (A) Weighing the curing agent and prepolymer of PDMS. (B) Pouring the mixed and deaerated mixture into a tape-patterned Petri dish. There were some newly generated air bubbles in the polymer mixture. (C) Deaerating the mixture again in a vacuum chamber. The air bubbles were rising and burst on the top surface. (D) The deaerated and cured PDMS resin. (E) Peeling off the cured PDMS elastomer from the Petri dish. (F) Cutting off every piece of PDMS channel blocks using a flat-cable cutter. (G) Punching holes at each end of the PDMS channel using a biopsy punch. (H) The assembled device. The PDMS resin can adhere to the CYTOP surface. (I) Translucent microchamber array area inside the PDMS channel filled with the aqueous solution before chilling. (J) Transparent microchamber array area inside the PDMS channel after chilling. Please click here to view a larger version of this figure.
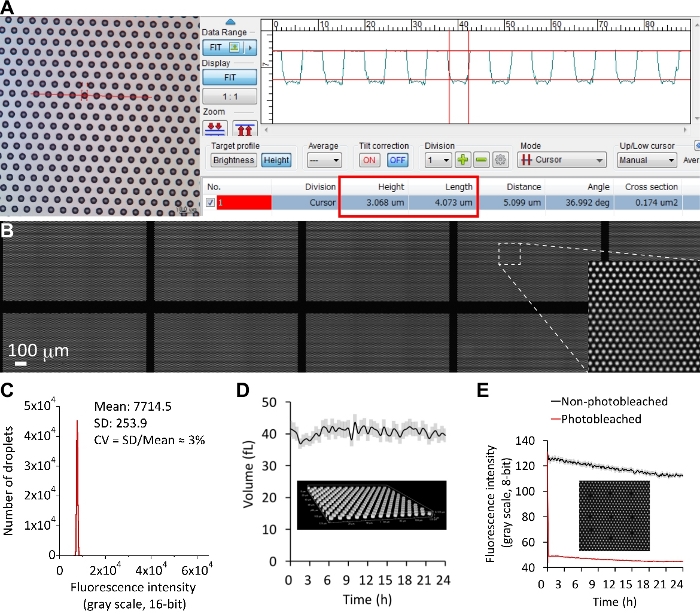
Figure 3: Ultra-uniform and ultra-stable femtoliter droplets. (A) 3D laser scanning confocal microscopy imaging for the fabricated substrate. The cylindrical microchambers in the example image showed a height of 3 μm and a diameter of 4 μm. (B) Uniform femtoliter droplets over a large area of the planar array. Only a partial area of the entire array was shown herein. A fluorescent solution (10 μM ATTO-514) was sealed in each droplet. (C) The size distribution of the droplets over an entire single array. The fluorescence intensity was used as an indicator of the droplet size. The CV was only 3%. (D) Volumetric measurement using confocal z-stack time-course data. The volume of droplets over the array was given by the microscope software (NIS-Elements, Nikon). (E) Fluorescence recovery after photobleaching. After the first frame, several droplets were completely photobleached using a confined laser beam of a confocal microscope. Their fluorescence intensity was recorded for 24 h, and no fluorescence recovery was observed (red line). The fluorescence intensity of other non-photobleached droplets in the same field of view was recorded in the black line. Error bars (translucent colors) were 1 SD for every time-point. Please click here to view a larger version of this figure.
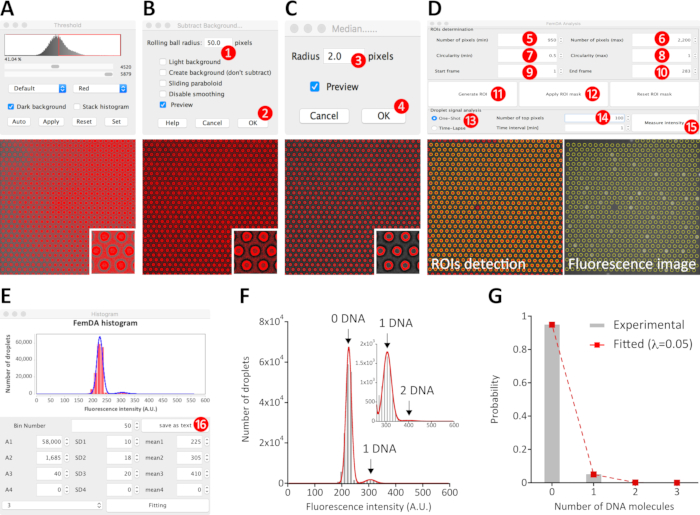
Figure 4: Analysis procedure of the end-point microscopic image data. The defocused bright-field image was used to extract the coordinate of every microchambers/droplets. Based on the intensity difference between the edge of microchambers (as foreground) and other areas (as background), the continuous and near-circular edge can be extracted from the background. Because of the uneven background distribution across the field of view (A), some image pre-processing is generally required to improve the quality of the binarizing output. After subtracting background (B), the background of every frame was uniformized. Empirically, the input value in the “Rolling ball radius” (step 1) was 20-50 for 2048 pixel × 2048 pixel images and 10-20 for 512 pixel × 512 pixel images. The larger the value, the shorter the processing time. To reduce the noise in the background-subtracted image, apply a median filter to the image (C). In general, the input value in the Radius (step 3) was 1-2 pixels. As shown in the magnified insert, the background-subtracted and filtered image can be nicely binarized using the build-in threshold plugin so that the foreground corresponding to the edges as well as the region of interests (ROIs) can be accurately recognized. The ROIs detection for all frames of the image was carried out by using the installed homemade plugin FemDA Analysis (D). The pixel size (steps 5 and 6) and circularity (steps 7 and 8) of ROIs, the frames that we want to put into the calculation (steps 9 and 10) were manually defined according to the actual data. After clicking Generate ROI (step 11) and waiting, the coordinate of every ROI across the input frames was determined. After clicking the Apply ROI mask (step 12), every detected ROI was enclosed and highlighted by a yellow line. After opening the fluorescence image and clicking the Apply ROI mask again, the determined ROIs mask was applied to the fluorescence image. The Number of top pixels (step 14) specified the number of top intensity-ranked pixels of each ROI for the calculation of the mean intensity of the respective droplets. After clicking Measure intensity (step 15) and waiting, the histogram was generated (E). The histogram can be fitted with a sum of Gaussian distributions using the parameters available in the histogram window. Alternatively, the mean intensity data can be exported to a text file (step 16) and analyzed by other software (F). (G) The probability of occurrence of droplets containing different numbers of DNA molecules in the given array. The histogram (grey color) was nicely fitted by a Poisson distribution (red dashed line) with an average of 0.05 DNA molecules per droplet, as expected for a random distribution of DNA molecules. Please click here to view a larger version of this figure.
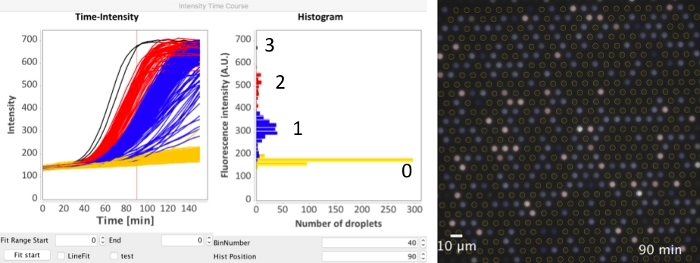
Figure 5: Analysis of the time-course microscopic image data. The detected ROIs (following the steps 1-12 of Figure 4) was directly applied to the time-course data. In step 13 of Figure 4, select Time-lapse and input the actual time-interval (in minutes) between adjacent frames in Time-interval [min]. After clicking Measure intensity (step 15 of Figure 4) and waiting, the time-intensity plot and the histogram (the same as Figure 4E) were generated. The Hist position specified the time-point of the histogram, which was shown as a vertical red line. The plugin also supports specifying colors for every trace line. Yellow: 0 DNA; blue was 1 DNA; red: 2 DNA; black: 3 DNA. The time-course data can also be exported to a text file. Please click here to view a larger version of this figure.
Supplementary Movie 1. Please click here to download this movie.
