RNA-seq ha sido ampliamente utilizado a lo largo de los años, generalmente para estimar la expresión génica diferencial y el descubrimiento de genes1. Además, también se puede utilizar para estimar el uso variable del nivel de exón debido a que los genes expresan diferentes isoformas, lo que contribuye a una mejor comprensión de la regulación génica a nivel post-transcripcional. La mayoría de los genes eucariotas generan diferentes isoformas mediante empalme alternativo (AS) para aumentar la diversidad de la expresión del ARNm. Los eventos AS se pueden dividir en diferentes patrones: salto de exones completos (SE) donde un exón (“casete”) se elimina completamente de la transcripción junto con sus intrones flanqueantes; selección alternativa (donante) del sitio de empalme de 5′ (A5SS) y selección alternativa 3′ (aceptor) del sitio de empalme (A3SS) cuando dos o más sitios de empalme están presentes en cada extremo de un exón; retención de intrones (RI) cuando un intrón se retiene dentro de la transcripción de ARNm maduro y exclusión mutua del uso de exones (MXE) donde solo uno de los dos exones disponibles puede ser retenido a la vez 2,3. La poliadenilación alternativa (APA) también juega un papel importante en la regulación de la expresión génica utilizando sitios alternativos de poli (A) para generar múltiples isoformas de ARNm a partir de una sola transcripción4. La mayoría de los sitios de poliadenilación (pAs) se encuentran en la región 3′ no traducida (3′ UTRs), generando isoformas de ARNm con diversas longitudes 3′ UTR. Como el 3′ UTR es el eje central para reconocer elementos reguladores, diferentes longitudes 3′ UTR pueden afectar la localización, estabilidad y traducción del ARNm5. Hay una clase de ensayos de secuenciación final 3′ optimizados para detectar APA que difieren en los detalles del protocolo6. La canalización descrita aquí está diseñada para PolyA-seq, pero se puede adaptar para otros protocolos como se describe.
En este estudio, presentamos una tubería de métodos de análisis de exones diferenciales7,8 (Figura 1), que se pueden dividir en dos grandes categorías: basados en exones (DEXSeq9, diffSplice10) y basados en eventos (replicate Multivariate Analysis of Transcript Splicing (rMATS)11). Los métodos basados en exones comparan el cambio de pliegue a través de las condiciones de los exones individuales, contra una medida del cambio general del pliegue genético para llamar al uso de exones expresado diferencialmente, y a partir de eso calcular una medida a nivel de gen de la actividad de AS. Los métodos basados en eventos utilizan lecturas de unión de expansión exón-intrón para detectar y clasificar eventos de empalme específicos, como la omisión de exón o la retención de intrones, y distinguir estos tipos de AS en la salida3. Por lo tanto, estos métodos proporcionan vistas complementarias para un análisis completo de la EA12,13. Se seleccionaron DEXSeq (basado en el paquete DESeq214 DGE) y diffSplice (basado en el paquete Limma10 DGE) para el estudio, ya que se encuentran entre los paquetes más utilizados para el análisis de empalme diferencial. rMATS fue elegido como un método popular para el análisis basado en eventos. Otro método popular basado en eventos es MISO (mezcla de isoformas)1. Para APA adaptamos el enfoque basado en exones.
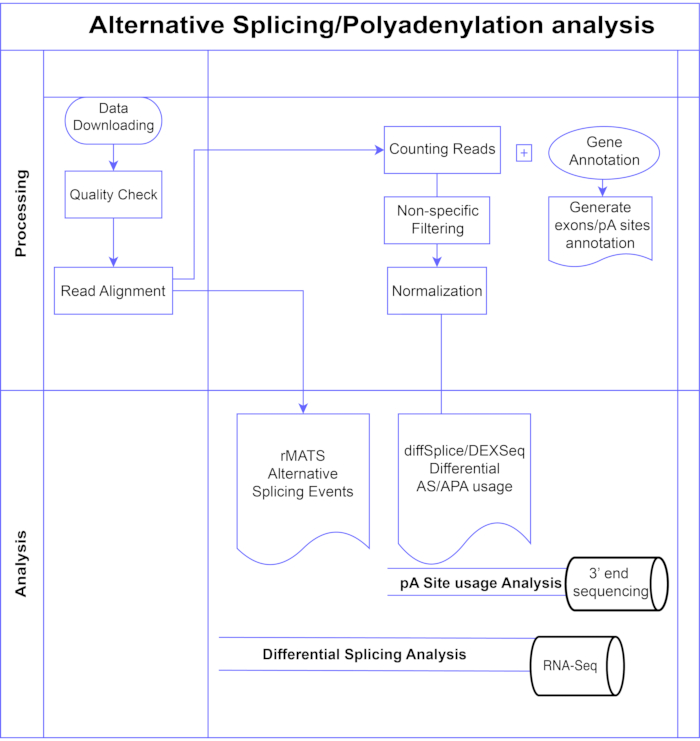
Figura 1. Pipeline de análisis. Diagrama de flujo de los pasos utilizados en el análisis. Los pasos incluyen: obtener los datos, realizar controles de calidad y alineación de lecturas seguidas de contar lecturas utilizando anotaciones para exones conocidos, intrones y sitios pA, filtrado para eliminar recuentos bajos y normalización. Los datos de PolyA-seq se analizaron para sitios de pA alternativos utilizando métodos diffSplice/DEXSeq, RNA-Seq a granel se analizaron para splicing alternativo a nivel de exón con métodos diffSplice/DEXseq, y los eventos de AS se analizaron con rMATS. Haga clic aquí para ver una versión más grande de esta figura.
Los datos de RNA-seq utilizados en este estudio fueron adquiridos de Gene Expression Omnibus (GEO) (GSE138691)15. Utilizamos datos de ARN-seq de ratón de este estudio con dos grupos de condiciones: tipo salvaje (WT) y knockout tipo 1 similar a Muscleblind (Mbnl1 KO) con tres réplicas cada uno. Para demostrar el análisis diferencial de uso del sitio de poliadenilación, obtuvimos datos de PolyA-seq de fibroblastos embrionarios de ratón (MEF) (GEO Accesion GSE60487)16. Los datos tienen cuatro grupos de condiciones: tipo salvaje (WT), tipo ciego muscular tipo 1 / tipo 2 doble knockout (Mbnl1/2 DKO), Mbnl 1/2 DKO con derribo Mbnl3 (KD) y Mbnl1/2 DKO con control Mbnl3 (Ctrl). Cada grupo de condición consta de dos réplicas.
| Adhesión al GEO | Número de ejecución de SRA | Nombre de la muestra | Condición | Replicar | Tejido | Secuenciación | Longitud de lectura | |
| RNA-Seq | GSM4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | Mbnl1 nocaut | Rep 1 | Timo | Extremo emparejado | 100 pb |
| GSM4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | Mbnl1 nocaut | Rep 2 | Timo | Extremo emparejado | 100 pb | |
| GSM4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | Mbnl1 nocaut | Rep 3 | Timo | Extremo emparejado | 100 pb | |
| GSM4116221 | SRR10261604 | WT_Thymus_1 | Tipo salvaje | Rep 1 | Timo | Extremo emparejado | 100 pb | |
| GSM4116222 | SRR10261605 | WT_Thymus_2 | Tipo salvaje | Rep 2 | Timo | Extremo emparejado | 100 pb | |
| GSM4116223 | SRR10261606 | WT_Thymus_3 | Tipo salvaje | Rep 3 | Timo | Extremo emparejado | 100 pb | |
| 3P-seq | GSM1480973 | SRR1553129 | WT_1 | Tipo salvaje (WT) | Rep 1 | Fibroblastos embrionarios de ratón (MEF) | Extremo único | 40 pb |
| GSM1480974 | SRR1553130 | WT_2 | Tipo salvaje (WT) | Rep 2 | Fibroblastos embrionarios de ratón (MEF) | Extremo único | 40 pb | |
| GSM1480975 | SRR1553131 | DKO_1 | Mbnl 1/2 doble knockout (DKO) | Rep 1 | Fibroblastos embrionarios de ratón (MEF) | Extremo único | 40 pb | |
| GSM1480976 | SRR1553132 | DKO_2 | Mbnl 1/2 doble knockout (DKO) | Rep 2 | Fibroblastos embrionarios de ratón (MEF) | Extremo único | 40 pb | |
| GSM1480977 | SRR1553133 | DKOsiRNA_1 | Mbnl 1/2 doble knockout con Mbnl 3 siRNA (KD) | Rep 1 | Fibroblastos embrionarios de ratón (MEF) | Extremo único | 40 pb | |
| GSM1480978 | SRR1553134 | DKOsiRNA_2 | Mbnl 1/2 doble knockout con Mbnl 3 siRNA (KD) | Rep 2 | Fibroblastos embrionarios de ratón (MEF) | Extremo único | 36 pb | |
| GSM1480979 | SRR1553135 | DKONTsiRNA_1 | Mbnl 1/2 doble knockout con siRNA no dirigido (Ctrl) | Rep 1 | Fibroblastos embrionarios de ratón (MEF) | Extremo único | 40 pb | |
| GSM1480980 | SRR1553136 | DKONTsiRNA_2 | Mbnl 1/2 doble knockout con siRNA no dirigido (Ctrl) | Rep 2 | Fibroblastos embrionarios de ratón (MEF) | Extremo único | 40 pb |
Tabla 1. Resumen de los conjuntos de datos RNA-Seq y PolyA-seq utilizados para el análisis.
