Le séquençage de l’ARN a été largement utilisé au fil des ans, généralement pour estimer l’expression différentielle des gènes et la découverte de gènes1. En outre, il peut également être utilisé pour estimer l’utilisation variable du niveau d’exon en raison de l’expression de gènes différentes isoformes, contribuant ainsi à une meilleure compréhension de la régulation des gènes au niveau post-transcriptionnel. La majorité des gènes eucaryotes génèrent différentes isoformes par épissage alternatif (AS) pour augmenter la diversité de l’expression de l’ARNm. Les événements AS peuvent être divisés en différents modèles: saut d’exons complets (SE) où un exon (« cassette ») est complètement retiré de la transcription avec ses introns flanquants; sélection alternative (donneur) du site d’épissage 5′ (A5SS) et alternative 3′ (accepteur) sélection du site d’épissage (A3SS) lorsque deux sites d’épissage ou plus sont présents à chaque extrémité d’un exon; rétention d’introns (RI) lorsqu’un intron est retenu dans le transcrit d’ARNm mature et exclusion mutuelle de l’utilisation d’exons (MXE) où un seul des deux exons disponibles peut être retenu à la fois 2,3. La polyadénylation alternative (APA) joue également un rôle important dans la régulation de l’expression génique en utilisant des sites poly (A) alternatifs pour générer plusieurs isoformes d’ARNm à partir d’un seul transcrit4. La plupart des sites de polyadénylation (pA) sont situés dans la région 3′ non traduite (3′ UTR), générant des isoformes d’ARNm avec diverses longueurs 3′ UTR. Comme l’UTR 3′ est le centre central pour la reconnaissance des éléments régulateurs, différentes longueurs 3′ UTR peuvent affecter la localisation, la stabilité et la traduction de l’ARNm5. Il existe une classe de tests de séquençage d’extrémité 3′ optimisés pour détecter l’APA qui diffèrent dans les détails du protocole6. Le pipeline décrit ici est conçu pour PolyA-seq, mais peut être adapté à d’autres protocoles comme décrit.
Dans cette étude, nous présentons un pipeline de méthodes d’analyse différentielledes exons 7,8 (Figure 1), qui peuvent être divisées en deux grandes catégories : basées sur les exons (DEXSeq9, diffSplice10) et basées sur les événements (analyse multivariée répliquée de l’épissage des transcriptions (rMATS)11). Les méthodes basées sur les exons comparent le changement de pli entre les conditions des exons individuels, à une mesure du changement global du pli des gènes pour appeler l’utilisation d’exons exprimée différentiellement, et à partir de là, calculez une mesure au niveau du gène de l’activité SA. Les méthodes basées sur les événements utilisent des lectures de jonction couvrant exon-intron pour détecter et classer des événements d’épissage spécifiques tels que le saut d’exon ou la rétention d’introns, et distinguer ces types AS dans la sortie3. Ainsi, ces méthodes fournissent des points de vue complémentaires pour une analyse complète de la SA12,13. Nous avons sélectionné DEXSeq (basé sur le package DESeq214 DGE) et diffSplice (basé sur le package Limma10 DGE) pour l’étude car ils sont parmi les packages les plus largement utilisés pour l’analyse d’épissage différentiel. rMATS a été choisi comme méthode populaire pour l’analyse basée sur les événements. Une autre méthode populaire basée sur les événements est MISO (Mixture of Isoforms)1. Pour l’APA, nous adaptons l’approche basée sur les exons.
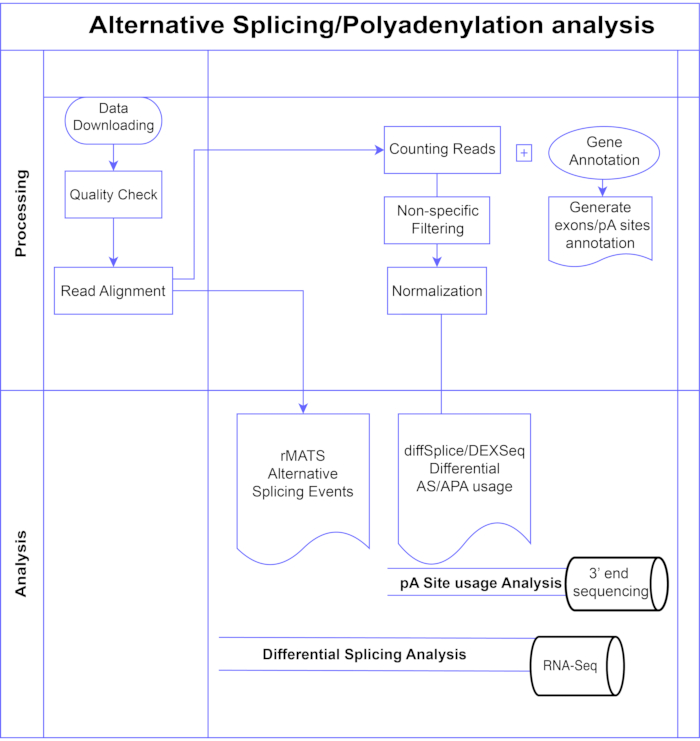
Graphique 1. Pipeline d’analyse. Organigramme des étapes utilisées dans l’analyse. Les étapes comprennent : l’obtention des données, l’exécution de contrôles de qualité et d’alignement des lectures, suivis du comptage des lectures à l’aide d’annotations pour les exons, les introns et les sites pA connus, le filtrage pour supprimer les faibles nombres et la normalisation. Les données PolyA-seq ont été analysées pour d’autres sites pA à l’aide des méthodes diffSplice/DEXSeq, le RNA-Seq en vrac a été analysé pour l’épissage alternatif au niveau de l’exon avec les méthodes diffSplice/DEXseq et les événements AS analysés avec rMATS. Veuillez cliquer ici pour voir une version agrandie de cette figure.
Les données RNA-seq utilisées dans cette enquête ont été acquises à partir de Gene Expression Omnibus (GEO) (GSE138691)15. Nous avons utilisé les données de séquençage de l’ARN de souris de cette étude avec deux groupes de conditions : le type sauvage (WT) et le knockout de type 1 de type Muscleblind (Mbnl1 KO) avec trois réplications chacun. Pour démontrer l’analyse différentielle de l’utilisation du site de polyadénylation, nous avons obtenu des données PolyA-seq sur des fibroblastes embryonnaires de souris (MEF) (GEO Accession GSE60487)16. Les données comportent quatre groupes de conditions : Wild-type (WT), Muscleblind-like type1/type 2 double knockout (Mbnl1/2 DKO), Mbnl 1/2 DKO avec Mbnl3 knockdown (KD) et Mbnl1/2 DKO avec contrôle Mbnl3 (Ctrl). Chaque groupe de conditions se compose de deux répétitions.
| Adhésion GEO | Numéro d’exécution SRA | Nom de l’échantillon | Condition | Répliquer | Tissu | Séquençage | Longueur de lecture | |
| RNA-Seq | GSM4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | Mbnl1 knockout | Rép. 1 | Thymus | Extrémité jumelée | 100 pb |
| GSM4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | Mbnl1 knockout | Rép. 2 | Thymus | Extrémité jumelée | 100 pb | |
| GSM4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | Mbnl1 knockout | Rép. 3 | Thymus | Extrémité jumelée | 100 pb | |
| GSM4116221 | SRR10261604 | WT_Thymus_1 | Type sauvage | Rép. 1 | Thymus | Extrémité jumelée | 100 pb | |
| GSM4116222 | SRR10261605 | WT_Thymus_2 | Type sauvage | Rép. 2 | Thymus | Extrémité jumelée | 100 pb | |
| GSM4116223 | SRR10261606 | WT_Thymus_3 | Type sauvage | Rép. 3 | Thymus | Extrémité jumelée | 100 pb | |
| 3P-Seq | GSM1480973 | SRR1553129 | WT_1 | Type sauvage (WT) | Rép. 1 | Fibroblastes embryonnaires de souris (MEF) | Extrémité unique | 40 pb |
| GSM1480974 | SRR1553130 | WT_2 | Type sauvage (WT) | Rép. 2 | Fibroblastes embryonnaires de souris (MEF) | Extrémité unique | 40 pb | |
| GSM1480975 | SRR1553131 | DKO_1 | Mbnl 1/2 double knockout (DKO) | Rép. 1 | Fibroblastes embryonnaires de souris (MEF) | Extrémité unique | 40 pb | |
| GSM1480976 | SRR1553132 | DKO_2 | Mbnl 1/2 double knockout (DKO) | Rép. 2 | Fibroblastes embryonnaires de souris (MEF) | Extrémité unique | 40 pb | |
| GSM1480977 | SRR1553133 | DKOsiRNA_1 | Mbnl 1/2 double knockout avec Mbnl 3 siRNA (KD) | Rép. 1 | Fibroblastes embryonnaires de souris (MEF) | Extrémité unique | 40 pb | |
| GSM1480978 | SRR1553134 | DKOsiRNA_2 | Mbnl 1/2 double knockout avec Mbnl 3 siRNA (KD) | Rép. 2 | Fibroblastes embryonnaires de souris (MEF) | Extrémité unique | 36 pb | |
| GSM1480979 | SRR1553135 | DKONTsiRNA_1 | Mbnl 1/2 double knockout avec siRNA non ciblé (Ctrl) | Rép. 1 | Fibroblastes embryonnaires de souris (MEF) | Extrémité unique | 40 pb | |
| GSM1480980 | SRR1553136 | DKONTsiRNA_2 | Mbnl 1/2 double knockout avec siRNA non ciblé (Ctrl) | Rép. 2 | Fibroblastes embryonnaires de souris (MEF) | Extrémité unique | 40 pb |
Tableau 1. Résumé des ensembles de données RNA-Seq et PolyA-seq utilisés pour l’analyse.
