多年来,RNA-seq已被广泛用于估计差异基因表达和基因发现1。此外,它还可用于估计由于表达不同亚型的基因而导致的不同外显子水平使用情况,从而有助于更好地了解转录后水平的基因调控。大多数真核基因通过交替剪接(AS)产生不同的亚型,以增加mRNA表达的多样性。AS事件可分为不同的模式:跳过完全外显子(SE),其中(“盒式”)外显子与其侧翼内含子一起从转录本中完全去除;当外显子两端存在两个或多个剪接位点时,备选(供体)5’剪接位点选择(A5SS)和备选方案3’(受体)剪接位点选择(A3SS);当内含子保留在成熟的mRNA转录本中时保留内含子(RI)和相互排除外显子使用(MXE),其中一次只能保留两个可用外显子中的一个2,3。替代聚腺苷酸化(APA)在使用替代聚(A)位点从单个转录本产生多种mRNA亚型4的基因表达中也起着重要作用。大多数聚腺苷酸化位点(pA)位于3’非翻译区域(3’UTR),产生具有不同3’UTR长度的mRNA亚型。由于 3′ UTR 是识别调控元件的中心枢纽,因此不同的 3′ UTR 长度会影响 mRNA 的定位、稳定性和翻译5。有一类 3′ 末端测序测定经过优化,可检测 APA,这些APA在协议6的细节上有所不同。此处描述的管道是为 PolyA-seq 设计的,但可以适用于所述的其他协议。
在这项研究中,我们提出了一系列差异外显子分析方法7,8 (图1),可分为两大类:基于外显子(DEXSeq9,diffSplice10)和基于事件的(复制转录本剪接的多变量分析(rMATS)11)。基于外显子的方法将单个外显子条件下的倍数变化与整体基因折叠变化的度量进行比较,以调用差异表达的外显子使用情况,并由此计算AS活性的基因水平测量。基于事件的方法使用外显子-内含子跨越结读取来检测和分类特定的剪接事件,例如外显子跳跃或内含子保留,并在输出3中区分这些AS类型。因此,这些方法为AS12,13的完整分析提供了补充观点。我们选择了DEXSeq(基于DESeq214 DGE封装)和diffSplice(基于Limma10 DGE封装)进行研究,因为它们是差分拼接分析中使用最广泛的软件包之一。rMATS被选为基于事件的分析的常用方法。另一种流行的基于事件的方法是MISO(亚型混合物)1。对于APA,我们采用基于外显子的方法。
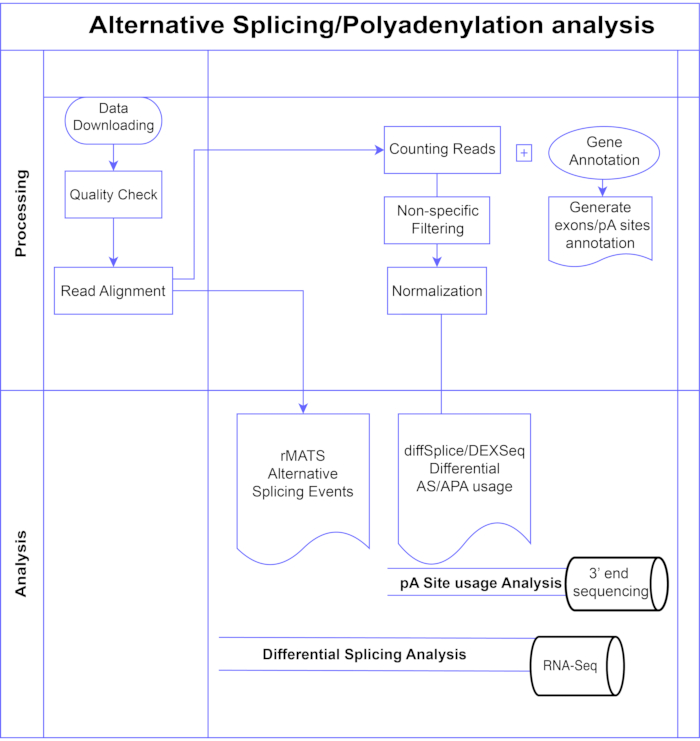
图1.分析管道。 分析中使用的步骤的流程图。步骤包括:获取数据,执行质量检查和读取对齐,然后使用已知外显子,内含子和pA位点的注释对读取进行计数,过滤以去除低计数和标准化。使用diffSplice/DEXSeq方法分析PolyA-seq数据以寻找替代pA位点,使用diffSplice/DEXseq方法分析外显子水平的替代剪接的体RNA-Seq,并使用rMATS分析AS事件。 请点击此处查看此图的大图。
本调查中使用的RNA-seq数据是从基因表达综合(GEO)(GSE138691)15获得的。我们使用本研究的小鼠RNA-seq数据,分为两个条件组:野生型(WT)和肌盲样1型敲除(Mbnl1 KO),每个重复三个。为了证明差异聚腺苷酸化位点的使用分析,我们获得了小鼠胚胎成纤维细胞(MEFs)PolyA-seq数据(GEO加入GSE60487)16。数据有四个条件组:野生型(WT),肌肉盲样1型/2型双敲除(Mbnl1 / 2 DKO),Mbnl 1 / 2 DKO与Mbnl3敲低(KD)和Mbnl1 / 2 DKO与Mbnl3对照(Ctrl)。每个条件组由两个仿行组成。
| 加入全球环境展望 | SRA 运行编号 | 示例名称 | 条件 | 复制 | 组织 | 测 序 | 读取长度 | |
| 核糖核酸序列 | GSM4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | Mbnl1 淘汰赛 | 代表 1 | 胸腺 | 配对端 | 100 基点 |
| GSM4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | Mbnl1 淘汰赛 | 代表 2 | 胸腺 | 配对端 | 100 基点 | |
| GSM4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | Mbnl1 淘汰赛 | 代表 3 | 胸腺 | 配对端 | 100 基点 | |
| GSM4116221 | SRR10261604 | WT_Thymus_1 | 野生型 | 代表 1 | 胸腺 | 配对端 | 100 基点 | |
| GSM4116222 | SRR10261605 | WT_Thymus_2 | 野生型 | 代表 2 | 胸腺 | 配对端 | 100 基点 | |
| GSM4116223 | SRR10261606 | WT_Thymus_3 | 野生型 | 代表 3 | 胸腺 | 配对端 | 100 基点 | |
| 3P-序列 | GSM1480973 | SRR1553129 | WT_1 | 野生型(WT) | 代表 1 | 小鼠胚胎成纤维细胞 (MEF) | 单端 | 40 基点 |
| GSM1480974 | SRR1553130 | WT_2 | 野生型(WT) | 代表 2 | 小鼠胚胎成纤维细胞 (MEF) | 单端 | 40 基点 | |
| GSM1480975 | SRR1553131 | DKO_1 | Mbnl 1/2 双淘汰赛 (DKO) | 代表 1 | 小鼠胚胎成纤维细胞 (MEF) | 单端 | 40 基点 | |
| GSM1480976 | SRR1553132 | DKO_2 | Mbnl 1/2 双淘汰赛 (DKO) | 代表 2 | 小鼠胚胎成纤维细胞 (MEF) | 单端 | 40 基点 | |
| GSM1480977 | SRR1553133 | DKOsiRNA_1 | Mbnl 1/2 双敲除与 Mbnl 3 siRNA (KD) | 代表 1 | 小鼠胚胎成纤维细胞 (MEF) | 单端 | 40 基点 | |
| GSM1480978 | SRR1553134 | DKOsiRNA_2 | Mbnl 1/2 双敲除与 Mbnl 3 siRNA (KD) | 代表 2 | 小鼠胚胎成纤维细胞 (MEF) | 单端 | 36 基点 | |
| GSM1480979 | SRR1553135 | DKONTsiRNA_1 | Mbnl 1/2 双敲除与非靶向 siRNA (Ctrl) | 代表 1 | 小鼠胚胎成纤维细胞 (MEF) | 单端 | 40 基点 | |
| GSM1480980 | SRR1553136 | DKONTsiRNA_2 | Mbnl 1/2 双敲除与非靶向 siRNA (Ctrl) | 代表 2 | 小鼠胚胎成纤维细胞 (MEF) | 单端 | 40 基点 |
表 1.用于分析的RNA-Seq和PolyA-seq数据集摘要。
