RNA-seq는 일반적으로 차등 유전자 발현 및유전자 발견을 추정하기 위해 수년에 걸쳐 널리 사용되어 왔습니다1. 또한 다양한 이소 형을 발현하는 유전자로 인해 다양한 엑손 수준 사용량을 추정하는 데 활용할 수 있으므로 전사 후 수준에서 유전자 조절을 더 잘 이해하는 데 기여할 수 있습니다. 대부분의 진핵생물 유전자는 mRNA 발현의 다양성을 증가시키기 위해 대안적 스플라이싱(AS)에 의해 상이한 이소형을 생성한다. AS 이벤트는 다른 패턴으로 나눌 수 있습니다 : ( “카세트”) 엑손이 측면 인트론과 함께 전사체에서 완전히 제거되는 완전한 엑손 (SE)의 건너 뛰기; 대안 (공여체) 5′ 스플라이스 부위 선택 (A5SS) 및 대안 3′ (수용체) 스플라이스 부위 선택 (A3SS) 2개 이상의 스플라이스 부위가 엑손의 양쪽 말단에 존재할 때; 인트론이 성숙한 mRNA 전사체 내에 유지되는 경우 인트론(RI)의 유지 및 한 번에 두 개의 사용 가능한 엑손 중 하나만 보유할 수 있는 엑손 사용(MXE)의 상호 배제 2,3. 대안적 폴리아데닐화 (APA)는 또한 단일 전사체로부터 다수의 mRNA 이소형을 생성하기 위해 대안적인 폴리 (A) 부위를 사용하여 유전자 발현을 조절하는데 중요한 역할을한다4. 대부분의 폴리아데닐화 부위 (pAs)는 3′ 비번역 영역 (3′ UTR) 내에 위치하여, 다양한 3′ UTR 길이를 갖는 mRNA 이소형을 생성한다. 3′ UTR이 조절 요소를 인식하기 위한 중앙 허브이기 때문에, 상이한 3′ UTR 길이는 mRNA 국소화, 안정성 및 번역(5)에 영향을 미칠 수 있다. 프로토콜6의 세부 사항에서 다른 APA를 검출하도록 최적화 된 3 ‘최종 시퀀싱 분석의 클래스가 있습니다. 여기에 설명된 파이프라인은 PolyA-seq용으로 설계되었지만 설명된 대로 다른 프로토콜에 맞게 조정할 수 있습니다.
이 연구에서는 차등 엑손 분석 방법7,8(그림 1)의 파이프라인을 제시하며, 이는 엑손 기반(DEXSeq9, diffSplice 10)과 이벤트 기반(전사체 접합의 복제 다변량 분석(rMATS)11)의 두 가지 범주로 나눌 수 있습니다. 엑손 기반 방법은 개별 엑손의 조건에 따른 폴드 변화를 차등적으로 발현된 엑손 사용량을 호출하기 위한 전체 유전자 폴드 변화의 척도와 비교하고, 그로부터 AS 활성의 유전자 수준 측정을 계산합니다. 이벤트 기반 방법은 엑손 인트론 스패닝 접합 읽기를 사용하여 엑손 건너뛰기 또는 인트론 유지와 같은 특정 스플라이싱 이벤트를 감지 및 분류하고 출력3에서 이러한 AS 유형을 구별합니다. 따라서 이러한 방법은 AS12,13의 완전한 분석을위한 보완적인 견해를 제공합니다. DEXSeq(DESeq214 DGE 패키지 기반)와 diffSplice(Limma10 DGE 패키지 기반)는 차동 접합 분석에 가장 널리 사용되는 패키지 중 하나이기 때문에 연구를 위해 선택했습니다. rMATS는 이벤트 기반 분석에 널리 사용되는 방법으로 선택되었습니다. 또 다른 인기 있는 이벤트 기반 방법은 MISO(Mix of Isoforms)1입니다. APA의 경우 엑손 기반 접근 방식을 적용합니다.
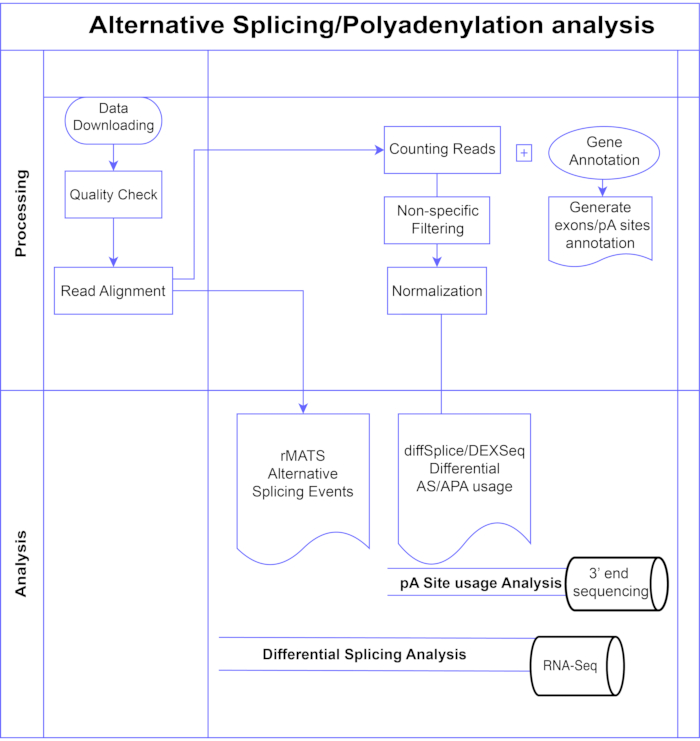
그림 1. 분석 파이프라인. 분석에 사용된 단계의 순서도입니다. 단계에는 데이터 획득, 품질 검사 수행 및 읽기 정렬 수행 후 알려진 엑손, 인트론 및 pA 사이트에 대한 주석을 사용하여 읽기 계산, 낮은 카운트 제거 및 정규화를 위한 필터링이 포함됩니다. PolyA-seq 데이터는 diffSplice/DEXSeq 방법을 사용하여 대체 pA 부위에 대해 분석되었고, 벌크 RNA-Seq는 diffSplice/DEXseq 방법을 사용하여 엑손 수준에서 대체 스플라이싱에 대해 분석되었으며, AS 이벤트는 rMATS로 분석되었습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
이 조사에 사용된 RNA-seq 데이터는 유전자 발현 옴니버스(GEO)(GSE138691)15에서 획득한 것이다. 우리는 이 연구의 마우스 RNA-seq 데이터를 야생형(WT) 및 근맹 유사 유형 1 녹아웃(Mbnl1 KO)의 두 가지 조건 그룹과 함께 각각 3개의 반복으로 사용했습니다. 차등 폴리아데닐화 부위 사용 분석을 입증하기 위해, 마우스 배아 섬유아세포(MEF) PolyA-seq 데이터(GEO Accession GSE60487)16를 얻었다. 데이터에는 야생형(WT), 근맹형 유형 1/유형 2 이중 녹아웃(Mbnl1/2 DKO), Mbnl3 녹다운(KD)이 있는 Mbnl 1/2 DKO 및 Mbnl3 대조군이 있는 Mbnl1/2 DKO(Ctrl)의 네 가지 조건 그룹이 있습니다. 각 조건 그룹은 두 번의 반복실험으로 구성됩니다.
| 지역 가입 | SRA 실행 번호 | 샘플 이름 | 조건 | 복제 | 조직 | 시퀀싱 | 읽기 길이 | |
| RNA-서열 | GSM4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | Mbnl1 녹아웃 | 담당자 1 | 흉선 | 페어링 엔드 | 100 bp |
| GSM4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | Mbnl1 녹아웃 | 담당자 2 | 흉선 | 페어링 엔드 | 100 bp | |
| GSM4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | Mbnl1 녹아웃 | 담당자 3 | 흉선 | 페어링 엔드 | 100 bp | |
| GSM4116221 | SRR10261604 | WT_Thymus_1 | 와일드 타입 | 담당자 1 | 흉선 | 페어링 엔드 | 100 bp | |
| GSM4116222 | SRR10261605 | WT_Thymus_2 | 와일드 타입 | 담당자 2 | 흉선 | 페어링 엔드 | 100 bp | |
| GSM4116223 | SRR10261606 | WT_Thymus_3 | 와일드 타입 | 담당자 3 | 흉선 | 페어링 엔드 | 100 bp | |
| 3P-시퀀스 | GSM1480973 | SRR1553129 | WT_1 | 와일드 타입 (WT) | 담당자 1 | 마우스 배아 섬유아세포(MEF) | 단일 종단 | 40 bp |
| GSM1480974 | SRR1553130 | WT_2 | 와일드 타입 (WT) | 담당자 2 | 마우스 배아 섬유아세포(MEF) | 단일 종단 | 40 bp | |
| GSM1480975 | SRR1553131 | DKO_1 | Mbnl 1/2 더블 녹아웃 (DKO) | 담당자 1 | 마우스 배아 섬유아세포(MEF) | 단일 종단 | 40 bp | |
| GSM1480976 | SRR1553132 | DKO_2 | Mbnl 1/2 더블 녹아웃 (DKO) | 담당자 2 | 마우스 배아 섬유아세포(MEF) | 단일 종단 | 40 bp | |
| GSM1480977 | SRR1553133 | DKOsiRNA_1 | Mbnl 3 siRNA (KD)를 사용한 Mbnl 1/2 이중 녹아웃 | 담당자 1 | 마우스 배아 섬유아세포(MEF) | 단일 종단 | 40 bp | |
| GSM1480978 | SRR1553134 | DKOsiRNA_2 | Mbnl 3 siRNA (KD)를 사용한 Mbnl 1/2 이중 녹아웃 | 담당자 2 | 마우스 배아 섬유아세포(MEF) | 단일 종단 | 36 bp | |
| GSM1480979 | SRR1553135 | DKONTsiRNA_1 | 비표적 siRNA를 사용한 Mbnl 1/2 이중 녹아웃(Ctrl) | 담당자 1 | 마우스 배아 섬유아세포(MEF) | 단일 종단 | 40 bp | |
| GSM1480980 | SRR1553136 | DKONTsiRNA_2 | 비표적 siRNA를 사용한 Mbnl 1/2 이중 녹아웃(Ctrl) | 담당자 2 | 마우스 배아 섬유아세포(MEF) | 단일 종단 | 40 bp |
표 1. 분석에 사용된 RNA-Seq 및 PolyA-seq 데이터 세트의 요약.
