RNA-seq har været meget udbredt gennem årene typisk til estimering af differentiel genekspression og genopdagelse1. Derudover kan det også bruges til at estimere varierende exon-niveau brug på grund af gen, der udtrykker forskellige isoformer, hvilket bidrager til en bedre forståelse af genregulering på post-transkriptionelt niveau. Størstedelen af eukaryote gener genererer forskellige isoformer ved alternativ splejsning (AS) for at øge mangfoldigheden af mRNA-ekspression. AS-begivenheder kan opdeles i forskellige mønstre: spring over komplette exons (SE), hvor en (“kassette”) exon fjernes helt ud af udskriften sammen med dens flankerende introns; alternativ (donor) 5′ splejsningssted (A5SS) og alternativ 3′ (acceptor) splejsningssted (A3SS), når to eller flere splejsningssteder er til stede i hver ende af en exon retention af introns (RI), når en intron bevares inden for det modne mRNA-transkript og gensidig udelukkelse af exonbrug (MXE), hvor kun en af de to tilgængelige exoner kan bevares ad gangen 2,3. Alternativ polyadenylering (APA) spiller også en vigtig rolle i reguleringen af genekspression ved hjælp af alternative poly (A) steder til at generere flere mRNA-isoformer fra et enkelt transkript4. De fleste polyadenyleringssteder (pA’er) er placeret i den 3′ uoversatte region (3′ UTR’er), der genererer mRNA-isoformer med forskellige 3′ UTR-længder. Da 3′ UTR er det centrale knudepunkt for genkendelse af regulatoriske elementer, kan forskellige 3′ UTR-længder påvirke mRNA-lokalisering, stabilitet og translation5. Der er en klasse af 3 ‘ende sekventeringsassays optimeret til at detektere APA, der adskiller sig i detaljerne i protokollen6. Rørledningen beskrevet her er designet til PolyA-seq, men kan tilpasses til andre protokoller som beskrevet.
I denne undersøgelse præsenterer vi en pipeline af differentielle exonanalysemetoder7,8 (figur 1), som kan opdeles i to brede kategorier: exonbaseret (DEXSeq9, diffSplice 10) og begivenhedsbaseret (replikeret multivariat analyse af transkriptionsplejsning (rMATS)11). De exon-baserede metoder sammenligner foldændringen på tværs af betingelser for individuelle exoner mod et mål for den samlede genfoldændring for at kalde differentielt udtrykt exon-brug og ud fra det beregne et mål på genniveau for AS-aktivitet. Hændelsesbaserede metoder bruger exon-intron-spanning junction-læsninger til at registrere og klassificere specifikke splejsningshændelser såsom exon-spring eller tilbageholdelse af introns og skelne mellem disse AS-typer i output3. Disse metoder giver således supplerende synspunkter til en komplet analyse af AS12,13. Vi valgte DEXSeq (baseret på DESeq214 DGE-pakken) og diffSplice (baseret på Limma10 DGE-pakken) til undersøgelsen, da de er blandt de mest anvendte pakker til differentiel splejsningsanalyse. rMATS blev valgt som en populær metode til begivenhedsbaseret analyse. En anden populær begivenhedsbaseret metode er MISO (Blanding af isoformer)1. For APA tilpasser vi den exon-baserede tilgang.
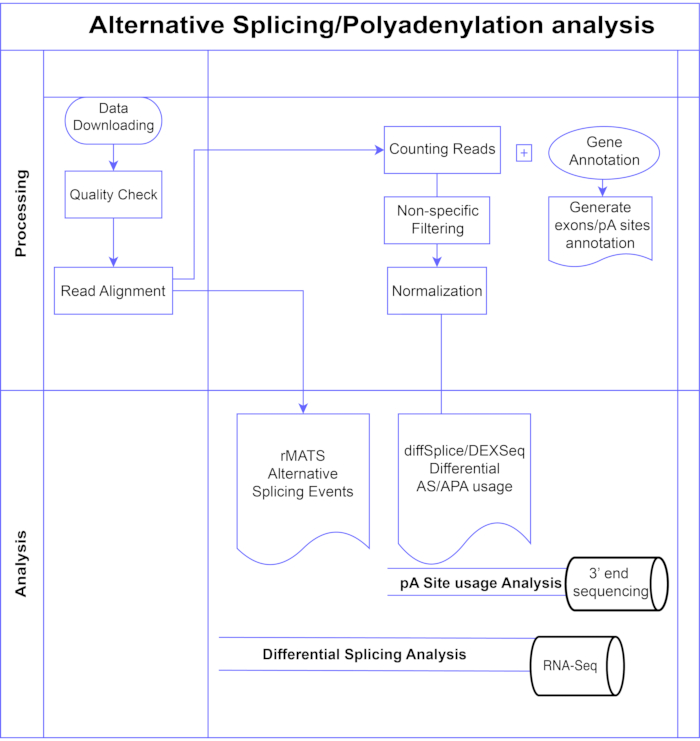
Figur 1. Analyse pipeline. Rutediagram over de trin, der bruges i analysen. Trin inkluderer: indhentning af data, udførelse af kvalitetskontrol og læsejustering efterfulgt af optælling af læsninger ved hjælp af kommentarer til kendte exons, introns og pA-websteder, filtrering for at fjerne lave tællinger og normalisering. PolyA-seq-data blev analyseret for alternative pA-steder ved hjælp af diffSplice/DEXSeq-metoder, bulk RNA-Seq blev analyseret for alternativ splejsning på exonniveau med diffSplice/DEXseq-metoder og AS-hændelser analyseret med rMATS. Klik her for at se en større version af denne figur.
RNA-seq-dataene, der blev brugt i denne undersøgelse, blev erhvervet fra Gene Expression Omnibus (GEO) (GSE138691)15. Vi brugte mus RNA-seq data fra denne undersøgelse med to tilstandsgrupper: wild-type (WT) og muskelblind-lignende type 1 knockout (Mbnl1 KO) med tre replikater hver. For at demonstrere differentiel analyse af brugen af polyadenyleringsstedet opnåede vi museembryofibroblaster (MEF’er) PolyA-seq-data (GEO-tiltrædelse GSE60487)16. Dataene har fire tilstandsgrupper: Wild-type (WT), Muscleblind-lignende type1/type 2 dobbelt knockout (Mbnl1/2 DKO), Mbnl 1/2 DKO med Mbnl3 knockdown (KD) og Mbnl1/2 DKO med Mbnl3 kontrol (Ctrl). Hver tilstandsgruppe består af to replikater.
| GEO tiltrædelse | SRA Kør nummer | Eksempel på navn | Betingelse | Kopiere | Væv | Sekventering | Læs længde | |
| RNA-Seq | GSM4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | Mbnl1 knockout | Rep 1 | Thymus | Parret ende | 100 bp |
| GSM4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | Mbnl1 knockout | Repræsentant 2 | Thymus | Parret ende | 100 bp | |
| GSM4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | Mbnl1 knockout | Rep 3 | Thymus | Parret ende | 100 bp | |
| GSM4116221 | SRR10261604 | WT_Thymus_1 | Vild type | Rep 1 | Thymus | Parret ende | 100 bp | |
| GSM4116222 | SRR10261605 | WT_Thymus_2 | Vild type | Repræsentant 2 | Thymus | Parret ende | 100 bp | |
| GSM4116223 | SRR10261606 | WT_Thymus_3 | Vild type | Rep 3 | Thymus | Parret ende | 100 bp | |
| 3P-Seq | GSM1480973 | SRR1553129 | WT_1 | Vildtype (WT) | Rep 1 | Museembryonale fibroblaster (MEF’er) | Single-end | 40 bp |
| GSM1480974 | SRR1553130 | WT_2 | Vildtype (WT) | Repræsentant 2 | Museembryonale fibroblaster (MEF’er) | Single-end | 40 bp | |
| GSM1480975 | SRR1553131 | DKO_1 | Mbnl 1/2 dobbelt knockout (DKO) | Rep 1 | Museembryonale fibroblaster (MEF’er) | Single-end | 40 bp | |
| GSM1480976 | SRR1553132 | DKO_2 | Mbnl 1/2 dobbelt knockout (DKO) | Repræsentant 2 | Museembryonale fibroblaster (MEF’er) | Single-end | 40 bp | |
| GSM1480977 | SRR1553133 | DKOsiRNA_1 | Mbnl 1/2 dobbelt knockout med Mbnl 3 siRNA (KD) | Rep 1 | Museembryonale fibroblaster (MEF’er) | Single-end | 40 bp | |
| GSM1480978 | SRR1553134 | DKOsiRNA_2 | Mbnl 1/2 dobbelt knockout med Mbnl 3 siRNA (KD) | Repræsentant 2 | Museembryonale fibroblaster (MEF’er) | Single-end | 36 bp | |
| GSM1480979 | SRR1553135 | DKONTsiRNA_1 | Mbnl 1/2 dobbelt knockout med ikke-målrettet siRNA (Ctrl) | Rep 1 | Museembryonale fibroblaster (MEF’er) | Single-end | 40 bp | |
| GSM1480980 | SRR1553136 | DKONTsiRNA_2 | Mbnl 1/2 dobbelt knockout med ikke-målrettet siRNA (Ctrl) | Repræsentant 2 | Museembryonale fibroblaster (MEF’er) | Single-end | 40 bp |
Tabel 1. Resumé af RNA-Seq og PolyA-seq datasæt, der anvendes til analysen.
