आरएनए-सेक का व्यापक रूप से वर्षों से व्यापक रूप से उपयोग किया गया है आमतौर पर अंतर जीन अभिव्यक्ति और जीन खोज1 का आकलन करने के लिए। इसके अलावा, इसका उपयोग विभिन्न आइसोफॉर्म व्यक्त करने वाले जीन के कारण अलग-अलग एक्सॉन स्तर के उपयोग का अनुमान लगाने के लिए भी किया जा सकता है, इसलिए पोस्ट-ट्रांसक्रिप्शनल स्तर पर जीन विनियमन की बेहतर समझ में योगदान देता है। यूकेरियोटिक जीन के बहुमत एमआरएनए अभिव्यक्ति की विविधता को बढ़ाने के लिए वैकल्पिक स्प्लिसिंग (एएस) द्वारा विभिन्न आइसोफॉर्म उत्पन्न करते हैं। एएस घटनाओं को विभिन्न पैटर्नों में विभाजित किया जा सकता है: पूर्ण एक्सॉन (एसई) को छोड़ना जहां एक (“कैसेट”) एक्सॉन को इसके फ्लैंकिंग इंट्रोन्स के साथ प्रतिलेख से पूरी तरह से हटा दिया जाता है; वैकल्पिक (दाता) 5 ‘स्प्लिस साइट चयन (ए 5 एसएस) और वैकल्पिक 3 ‘ (स्वीकर्ता) स्प्लिस साइट चयन (ए 3 एसएस) जब एक्सॉन के दोनों छोर पर दो या दो से अधिक स्प्लिस साइटें मौजूद होती हैं; इंट्रोन्स (आरआई) का प्रतिधारण जब एक इंट्रोन को परिपक्व एमआरएनए प्रतिलेख और एक्सॉन उपयोग (एमएक्सई) के पारस्परिक बहिष्करण के भीतर बनाए रखा जाता है, जहां दो उपलब्ध एक्सॉन में से केवल एक कोएक समय में बनाए रखा जा सकता है। वैकल्पिक पॉलीएडेनाइलेशन (एपीए) एक एकल प्रतिलेख4 से कई एमआरएनए आइसोफॉर्म उत्पन्न करने के लिए वैकल्पिक पॉली (ए) साइटों का उपयोग करके जीन अभिव्यक्ति को विनियमित करने में भी महत्वपूर्ण भूमिका निभाता है। अधिकांश पॉलीएडेनाइलेशन साइटें (पीए) 3 ‘अअनुवादित क्षेत्र (3’ यूटीआर) में स्थित हैं, जो विविध 3 ‘ यूटीआर लंबाई के साथ एमआरएनए आइसोफॉर्म उत्पन्न करती हैं। चूंकि 3 ‘यूटीआर नियामक तत्वों को पहचानने के लिए केंद्रीय केंद्र है, इसलिए विभिन्न 3 ‘यूटीआर लंबाई एमआरएनए स्थानीयकरण, स्थिरता और अनुवाद5 को प्रभावित कर सकती है। एपीए का पता लगाने के लिए अनुकूलित 3 ‘अंत अनुक्रमण परखों का एक वर्ग है जो प्रोटोकॉल6 के विवरण में भिन्न है। यहां वर्णित पाइपलाइन पॉलीए-सेक के लिए डिज़ाइन की गई है, लेकिन वर्णित अन्य प्रोटोकॉल के लिए अनुकूलित किया जा सकता है।
इस अध्ययन में, हम विभेदक एक्सॉन विश्लेषण विधियों 7,8 (चित्रा 1) की एक पाइपलाइन प्रस्तुत करते हैं, जिसे दो व्यापक श्रेणियों में विभाजित किया जा सकता है: एक्सॉन-आधारित (DEXSeq9, डिफस्पिस्प्लिस10) और इवेंट-आधारित (ट्रांसक्रिप्ट स्प्लिसिंग के मल्टीवेरिएट विश्लेषण (rMATS)11 को दोहराएं)। एक्सॉन-आधारित विधियां अलग-अलग एक्सॉन की स्थितियों में गुना परिवर्तन की तुलना करती हैं, समग्र जीन फोल्ड परिवर्तन के माप के खिलाफ अलग-अलग व्यक्त एक्सॉन उपयोग को कॉल करती हैं, और इससे एएस गतिविधि के जीन-स्तर माप की गणना करती हैं। इवेंट-आधारित विधियां एक्सॉन-इंट्रॉन-पैनिंग जंक्शन रीड का उपयोग विशिष्ट स्प्लिसिंग घटनाओं का पता लगाने और वर्गीकृत करने के लिए करती हैं जैसे कि एक्सॉन स्किपिंग या इंट्रोन्स के प्रतिधारण, और आउटपुट3 में इन एएस प्रकारों को अलग करना। इस प्रकार, ये विधियां एएस12,13 के पूर्ण विश्लेषण के लिए पूरक विचार प्रदान करती हैं। हमने अध्ययन के लिए DEXSeq (DESeq214 DGE पैकेज के आधार पर) और डिफस्प्लिस (लिम्मा10 DGE पैकेज के आधार पर) का चयन किया क्योंकि वे विभेदक स्प्लिसिंग विश्लेषण के लिए सबसे व्यापक रूप से उपयोग किए जाने वाले पैकेजों में से हैं। आरएमएटीएस को घटना-आधारित विश्लेषण के लिए एक लोकप्रिय विधि के रूप में चुना गया था। एक और लोकप्रिय घटना-आधारित विधि एमआईएसओ (आइसोफॉर्म का मिश्रण) 1 है। एपीए के लिए हम एक्सॉन-आधारित दृष्टिकोण को अनुकूलित करते हैं।
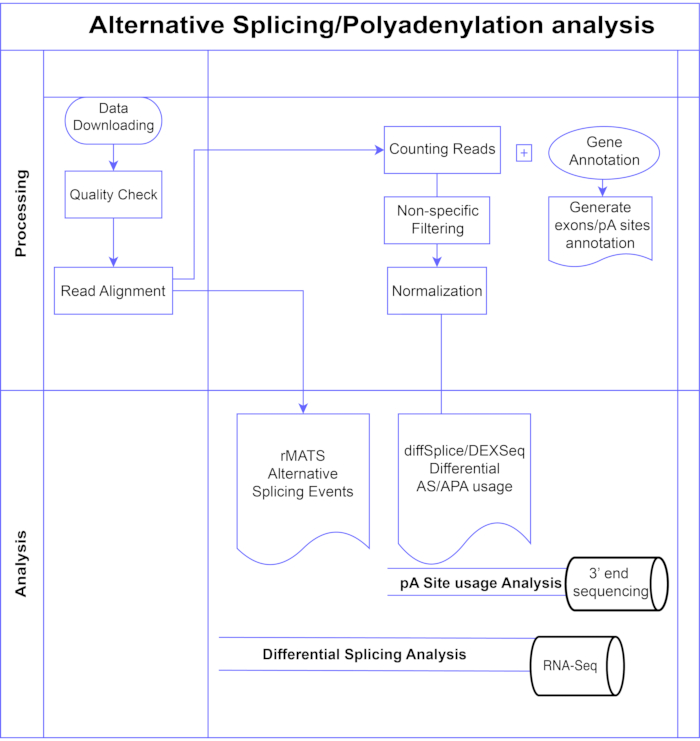
चित्र 1. विश्लेषण पाइपलाइन। विश्लेषण में उपयोग किए गए चरणों का फ़्लोचार्ट। चरणों में शामिल हैं: डेटा प्राप्त करना, गुणवत्ता जांच करना और संरेखण पढ़ना, जिसके बाद ज्ञात एक्सॉन, इंट्रोन्स और पीए साइटों के लिए एनोटेशन का उपयोग करके रीड की गिनती, कम गिनती को हटाने और सामान्यीकरण के लिए फ़िल्टर करना। डिफस्प्लिस/डीईएक्ससेक विधियों का उपयोग करके वैकल्पिक पीए साइटों के लिए पॉलीए-सेक डेटा का विश्लेषण किया गया था, थोक आरएनए-सेक का विश्लेषण एक्सॉन स्तर पर वैकल्पिक स्प्लिसिंग के लिए डिफस्प्लिस / डीईएक्ससेक विधियों के साथ किया गया था, और एएस घटनाओं का विश्लेषण आरएमएटीएस के साथ किया गया था। कृपया इस आंकड़े का एक बड़ा संस्करण देखने के लिए यहां क्लिक करें।
इस सर्वेक्षण में उपयोग किए गए आरएनए-सेक डेटा को जीन एक्सप्रेशन ओमनीबस (जीईओ) (जीएसई 138691)15 से प्राप्त किया गया था। हमने इस अध्ययन से माउस आरएनए-सेक डेटा का उपयोग दो स्थिति समूहों के साथ किया: वाइल्ड-टाइप (डब्ल्यूटी) और मसलब्लाइंड जैसे टाइप 1 नॉकआउट (एमबीएनएल 1 केओ) प्रत्येक में तीन प्रतिकृतियां थीं। विभेदक पॉलीएडेनाइलेशन साइट उपयोग विश्लेषण का प्रदर्शन करने के लिए, हमने माउस भ्रूण फाइब्रोब्लास्ट (एमईएफ) पॉलीए-सेक डेटा (जीईओ परिग्रहण जीएसई 60487)16 प्राप्त किया। डेटा में चार स्थिति समूह हैं: वाइल्ड-टाइप (डब्ल्यूटी), मसलब्लाइंड जैसे टाइप 1/टाइप 2 डबल नॉकआउट (एमबीएनएल 1/2 डीकेओ), एमबीएनएल 1/2 डीकेओ के साथ एमबीएनएल 3 वध (केडी) और एमबीएनएल 3 नियंत्रण (सीटीआरएल) के साथ एमबीएनएल 1/2 डीकेओ। प्रत्येक स्थिति समूह में दो प्रतिकृतियां होती हैं।
| जीईओ परिग्रहण | SRA रन नंबर | नमूना नाम | दशा | प्रतिकृति | ऊतक | अनुक्रमण | पढ़ने की लंबाई | |
| आरएनए-सेक | GSM4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | Mbnl1 नॉकआउट | प्रतिनिधि 1 | थाइमस | पेयर-एंड | 100 bp |
| GSM4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | Mbnl1 नॉकआउट | प्रतिनिधि 2 | थाइमस | पेयर-एंड | 100 bp | |
| GSM4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | Mbnl1 नॉकआउट | प्रतिनिधि 3 | थाइमस | पेयर-एंड | 100 bp | |
| GSM4116221 | SRR10261604 | WT_Thymus_1 | जंगली प्रकार | प्रतिनिधि 1 | थाइमस | पेयर-एंड | 100 bp | |
| GSM4116222 | SRR10261605 | WT_Thymus_2 | जंगली प्रकार | प्रतिनिधि 2 | थाइमस | पेयर-एंड | 100 bp | |
| GSM4116223 | SRR10261606 | WT_Thymus_3 | जंगली प्रकार | प्रतिनिधि 3 | थाइमस | पेयर-एंड | 100 bp | |
| 3P-Seq | GSM1480973 | SRR1553129 | WT_1 | जंगली प्रकार (WT) | प्रतिनिधि 1 | माउस भ्रूण फाइब्रोब्लास्ट (एमईएफ) | सिंगल-एंड | 40 बीपी |
| GSM1480974 | SRR1553130 | WT_2 | जंगली प्रकार (WT) | प्रतिनिधि 2 | माउस भ्रूण फाइब्रोब्लास्ट (एमईएफ) | सिंगल-एंड | 40 बीपी | |
| GSM1480975 | SRR1553131 | DKO_1 | एमबीएनएल 1/2 डबल नॉकआउट (डीकेओ) | प्रतिनिधि 1 | माउस भ्रूण फाइब्रोब्लास्ट (एमईएफ) | सिंगल-एंड | 40 बीपी | |
| GSM1480976 | SRR1553132 | DKO_2 | एमबीएनएल 1/2 डबल नॉकआउट (डीकेओ) | प्रतिनिधि 2 | माउस भ्रूण फाइब्रोब्लास्ट (एमईएफ) | सिंगल-एंड | 40 बीपी | |
| GSM1480977 | SRR1553133 | DKOsiRNA_1 | Mbnl 3 sirna (KD) के साथ Mbnl 1/2 डबल नॉकआउट | प्रतिनिधि 1 | माउस भ्रूण फाइब्रोब्लास्ट (एमईएफ) | सिंगल-एंड | 40 बीपी | |
| GSM1480978 | SRR1553134 | DKOsiRNA_2 | Mbnl 3 sirna (KD) के साथ Mbnl 1/2 डबल नॉकआउट | प्रतिनिधि 2 | माउस भ्रूण फाइब्रोब्लास्ट (एमईएफ) | सिंगल-एंड | 36 बीपी | |
| GSM1480979 | SRR1553135 | DKONTsiRNA_1 | गैर-लक्ष्यीकरण सीआरएनए (Ctrl) के साथ Mbnl 1/2 डबल नॉकआउट | प्रतिनिधि 1 | माउस भ्रूण फाइब्रोब्लास्ट (एमईएफ) | सिंगल-एंड | 40 बीपी | |
| GSM1480980 | SRR1553136 | DKONTsiRNA_2 | गैर-लक्ष्यीकरण सीआरएनए (Ctrl) के साथ Mbnl 1/2 डबल नॉकआउट | प्रतिनिधि 2 | माउस भ्रूण फाइब्रोब्लास्ट (एमईएफ) | सिंगल-एंड | 40 बीपी |
तालिका 1. विश्लेषण के लिए उपयोग किए जाने वाले आरएनए-सेक और पॉलीए-सेक डेटासेट का सारांश।
