RNA-seq har blitt mye brukt gjennom årene, typisk for å estimere differensial genuttrykk og genoppdagelse1. I tillegg kan det også brukes til å estimere varierende eksonnivåbruk på grunn av genuttrykk for forskjellige isoformer, og dermed bidra til en bedre forståelse av genregulering på posttranskripsjonsnivå. Flertallet av eukaryote gener genererer forskjellige isoformer ved alternativ spleising (AS) for å øke mangfoldet av mRNA-uttrykk. AS-hendelser kan deles inn i forskjellige mønstre: hopping av komplette eksoner (SE) der en (“kassett”) ekson fjernes helt ut av transkripsjonen sammen med dens flankerende introner; alternativ (donor) 5′ valg av spleisested (A5SS) og alternativ 3′ (akseptor) spleisestedsvalg (A3SS) når to eller flere skjøtesteder er til stede i hver ende av en ekson; oppbevaring av introner (RI) når et intron beholdes i det modne mRNA-transkriptet og gjensidig utelukkelse av eksonbruk (MXE) der bare ett av de to tilgjengelige eksonene kan beholdes om gangen 2,3. Alternativ polyadenylering (APA) spiller også en viktig rolle i regulering av genuttrykk ved bruk av alternative poly (A) steder for å generere flere mRNA-isoformer fra et enkelt transkripsjon4. De fleste polyadenyleringssteder (pAs) er lokalisert i 3 ‘uoversatte regionen (3’ UTRs), og genererer mRNA-isoformer med forskjellige 3 ‘UTR-lengder. Siden 3′ UTR er det sentrale knutepunktet for å gjenkjenne regulatoriske elementer, kan forskjellige 3’ UTR-lengder påvirke mRNA-lokalisering, stabilitet og oversettelse5. Det er en klasse med 3 ‘endesekvenseringsanalyser optimalisert for å oppdage APA som er forskjellige i detaljene i protokollen6. Rørledningen beskrevet her er designet for PolyA-seq, men kan tilpasses for andre protokoller som beskrevet.
I denne studien presenterer vi en pipeline av differensielle eksonanalysemetoder7,8 (figur 1), som kan deles inn i to brede kategorier: eksonbasert (DEXSeq9, diffSplice 10) og hendelsesbasert (replikat multivariat analyse av transkripsjonsspleising (rMATS)11). De eksonbaserte metodene sammenligner foldeendringen på tvers av forholdene til individuelle eksoner, mot et mål på total genfoldendring for å kalle differensielt uttrykt eksonbruk, og fra det beregnes et gennivåmål for AS-aktivitet. Hendelsesbaserte metoder bruker ekson-intron-spennende kryssavlesninger for å oppdage og klassifisere spesifikke spleisehendelser som eksonhopping eller oppbevaring av introner, og skille disse AS-typene i utgangen3. Dermed gir disse metodene komplementære visninger for en fullstendig analyse av AS12,13. Vi valgte DEXSeq (basert på DESeq214 DGE-pakken) og diffSplice (basert på Limma10 DGE-pakken) for studien, da de er blant de mest brukte pakkene for differensiell spleisingsanalyse. rMATS ble valgt som en populær metode for hendelsesbasert analyse. En annen populær hendelsesbasert metode er MISO (Blanding av isoformer)1. For TFO tilpasser vi den eksonbaserte tilnærmingen.
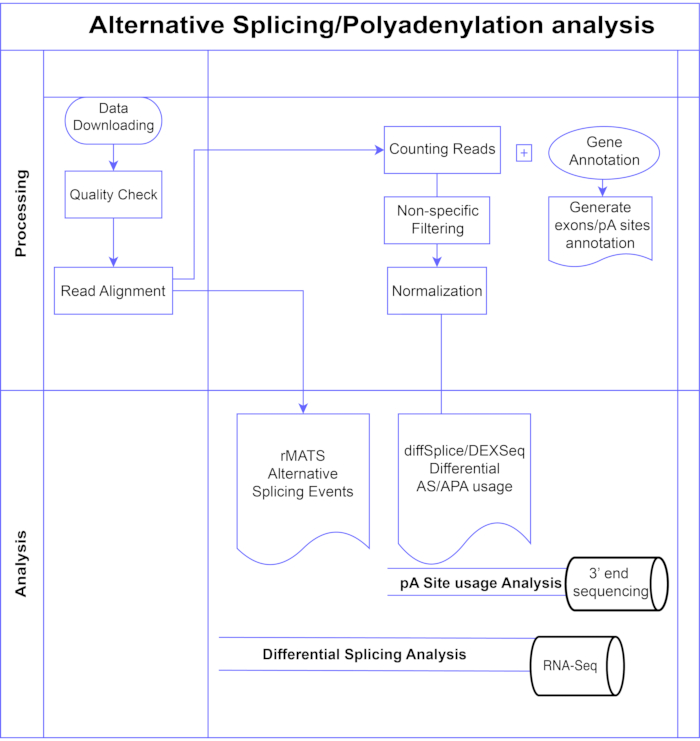
Figur 1. Analyse rørledning. Flytskjema for trinnene som brukes i analysen. Trinn inkluderer: innhenting av data, utførelse av kvalitetskontroller og lesejustering etterfulgt av telling av avlesninger ved hjelp av merknader for kjente eksoner, introner og pA-nettsteder, filtrering for å fjerne lave tellinger og normalisering. PolyA-seq-data ble analysert for alternative pA-lokaliteter med diffSplice/DEXSeq-metoder, bulk RNA-Seq ble analysert for alternativ spleising på eksonnivå med diffSplice/DEXseq-metoder, og AS-hendelser analysert med rMATS. Vennligst klikk her for å se en større versjon av denne figuren.
RNA-seq-dataene som ble brukt i denne undersøkelsen ble hentet fra Gene Expression Omnibus (GEO) (GSE138691)15. Vi brukte mus RNA-seq data fra denne studien med to tilstandsgrupper: villtype (WT) og Muskelblind-lignende type 1 knockout (Mbnl1 KO) med tre replikasjoner hver. For å demonstrere differensiell analyse av bruk av polyadenyleringsstedet, oppnådde vi museembryofibroblaster (MEFs) PolyA-seq-data (GEO Accession GSE60487)16. Dataene har fire tilstandsgrupper: Wild-type (WT), Muscleblind-lignende type1/type 2 dobbel knockout (Mbnl1/2 DKO), Mbnl 1/2 DKO med Mbnl3 knockdown (KD) og Mbnl1/2 DKO med Mbnl3 kontroll (Ctrl). Hver tilstandsgruppe består av to replikaer.
| GEO-tiltredelse | SRA Run nummer | Eksempel på navn | Betingelse | Replikere | Vev | Sekvensering | Lese lengde | |
| RNA-Seq | GSM4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | Mbnl1 knockout | Rep 1 | Thymus | Par-ende | 100 bp |
| GSM4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | Mbnl1 knockout | Rep 2 | Thymus | Par-ende | 100 bp | |
| GSM4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | Mbnl1 knockout | Rep 3 | Thymus | Par-ende | 100 bp | |
| GSM4116221 | SRR10261604 | WT_Thymus_1 | Vill type | Rep 1 | Thymus | Par-ende | 100 bp | |
| GSM4116222 | SRR10261605 | WT_Thymus_2 | Vill type | Rep 2 | Thymus | Par-ende | 100 bp | |
| GSM4116223 | SRR10261606 | WT_Thymus_3 | Vill type | Rep 3 | Thymus | Par-ende | 100 bp | |
| 3P-Seq | GSM1480973 | SRR1553129 | WT_1 | Vill type (WT) | Rep 1 | Mus embryonale fibroblaster (MEFs) | Single-end | 40 bp |
| GSM1480974 | SRR1553130 | WT_2 | Vill type (WT) | Rep 2 | Mus embryonale fibroblaster (MEFs) | Single-end | 40 bp | |
| GSM1480975 | SRR1553131 | DKO_1 | Mbnl 1/2 dobbel knockout (DKO) | Rep 1 | Mus embryonale fibroblaster (MEFs) | Single-end | 40 bp | |
| GSM1480976 | SRR1553132 | DKO_2 | Mbnl 1/2 dobbel knockout (DKO) | Rep 2 | Mus embryonale fibroblaster (MEFs) | Single-end | 40 bp | |
| GSM1480977 | SRR1553133 | DKOsiRNA_1 | Mbnl 1/2 dobbel knockout med Mbnl 3 siRNA (KD) | Rep 1 | Mus embryonale fibroblaster (MEFs) | Single-end | 40 bp | |
| GSM1480978 | SRR1553134 | DKOsiRNA_2 | Mbnl 1/2 dobbel knockout med Mbnl 3 siRNA (KD) | Rep 2 | Mus embryonale fibroblaster (MEFs) | Single-end | 36 bp | |
| GSM1480979 | SRR1553135 | DKONTsiRNA_1 | Mbnl 1/2 dobbel knockout med ikke-målretting siRNA (Ctrl) | Rep 1 | Mus embryonale fibroblaster (MEFs) | Single-end | 40 bp | |
| GSM1480980 | SRR1553136 | DKONTsiRNA_2 | Mbnl 1/2 dobbel knockout med ikke-målretting siRNA (Ctrl) | Rep 2 | Mus embryonale fibroblaster (MEFs) | Single-end | 40 bp |
Tabell 1. Sammendrag av RNA-Seq og PolyA-seq datasett som brukes til analysen.
