Vores forståelse af celledifferentiering og vævs og organers tilblivelse er resultatet af årtiers detaljerede målrettede screeninger af gener og deres produkter. At øge vores viden om alle biomolekylerne og deres mængder under vigtige cellulære begivenheder ville hjælpe med at optrævle molekylære mekanismer, der styrer det rumlige og tidsmæssige mønster af hvirveldyrs kropsplan. Teknologier, der muliggør molekylær amplifikation og sekventering, er nu i stand til rutinemæssigt at rapportere om et stort antal gener og transkripter, hvilket understøtter hypotesedrevne undersøgelser inden for grundlæggende biologisk og translationel forskning. For at forstå udviklingssystemer går et komplekst forhold mellem transkription og oversættelse ind for direkte analyse af flere proteiner og deres posttranslationelle modifikationer. Global proteomics ved hjælp af in vitro biologiske systemer, såsom inducerede pluripotente stamceller, begyndte at afgrænse mekanismer for vævsinduktion 1,2. I komplekse organismer, såsom hvirveldyrembryoet, afhænger udviklingen af morfogengradienter i sammenhæng med rum og tid3. Det følger heraf, at det at få viden om proteomiske ændringer, når celler differentierer sig til dannelse af specialiserede væv, såsom neurale væv, giver en nøgle til at låse op for molekylære programmer, der styrer normal og defekt udvikling og guide næste generations terapi.
Hvirveldyret sydafrikansk klofrø (Xenopus laevis) er en veletableret model inden for celle- og udviklings-, neuro- og regenerativ biologi. Sir John Gurdons Nobelpris i fysiologi eller medicin 4,5 i 2012 for opdagelsen af pluripotens af den somatiske kerne fremhævede betydningen af denne model for opdagelser i grundlæggende og translationelle studier. Xenopus-embryoner udvikler sig eksternt til moderen, hvilket letter direkte manipulation af celler, cellekloner og genekspression over forskellige udviklingsstadier. Asymmetrisk pigmentering og stereotype celledelinger gjorde det muligt at kortlægge reproducerbare skæbnekort fra 16-6 og 32-celle 7,8-stadieembryoet. For proteomics baseret på massespektrometri med høj opløsning (HRMS) omfatter yderligere fordele ved modellen relativt stor størrelse (~1 mm i diameter), hvilket giver rigeligt proteinindhold til analyse (~130 μg i embryoner i tidlig spaltningsfase, ~10 μg proteinindhold i enkeltceller i det 16-cellede embryo)9,10.
På nuværende tidspunkt er HRMS den førende foretrukne teknologi til påvisning af proteiner. Denne teknologi muliggør direkte, følsom og specifik detektion og kvantificering af flere, normalt hundreder til tusinder af forskellige proteiner11. Bottom-up proteomics af HRMS involverer en række indbyrdes forbundne trin. Efter ekstraktion fra celle/vævsprøven fordøjes proteiner med et proteolytisk enzym, såsom trypsin (bottom-up proteomics). De resulterende peptider adskilles baseret på deres forskellige fysisk-kemiske egenskaber, herunder hydrofobicitet (omvendt fase væskekromatografi, LC), nettoladning (ionbytterkromatografi), størrelse (størrelseseksklusionskromatografi) eller elektroforetisk mobilitet (kapillærelektroforese, CE). Peptider lades derefter (ioniseres), typisk ved hjælp af elektrosprayionisering (ESI), og peptidioner detekteres og sekventeres via gasfasefragmentering ved tandem HRMS. De resulterende peptiddata kortlægges til proteomet af organismen, der undersøges. Med proteinspecifik (proteotypisk) peptidionsignalintensitet, der korrelerer med koncentration, kan proteinkvantificering udføres etiketfri eller etiketbaseret (multiplexingkvantificering). HRMS proteomics giver en rig ressource af information om den molekylære tilstand af det undersøgte system, hvilket giver mulighed for generering af hypoteser og opfølgende funktionelle undersøgelser.
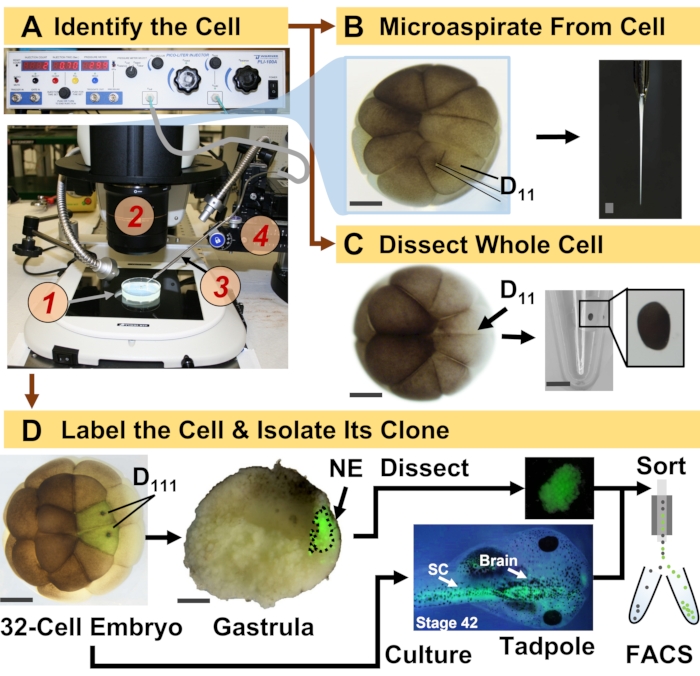
Figur 1: Spatiotemporalt skalerbar proteomics, der muliggør celleafstamningsstyret HRMS-proteomics i det udviklende (frø) embryo. (A) Visualisering af prøven (1) ved hjælp af et stereomikroskop (2) til injektion af en identificeret celle (indsats) ved hjælp af en fabrikeret mikropipette (3) under kontrol ved hjælp af et translationstrin (4). B) Subcellulær prøveudtagning af den identificerede venstre D11-celle i et 16-cellet embryo. C) Dissektion af en hel D11-celle fra et 16-cellet embryo. (D) Fluorescerende (grøn) sporing af venstre og højre D111-afkom fra et 32-cellet embryo for at styre dissektion af neurale ektoderm (NE) i gastrula (trin 10) og isolering af det nedadgående væv fra haletudsen ved hjælp af FACS. Skalastænger: 200 μm for embryoner, 1,25 mm for hætteglasset. Tallene er tilpasset med tilladelse fra referencerne 15,19,21,59. Klik her for at se en større version af denne figur.
Den protokol, der præsenteres her, muliggør HRMS-baseret kvantificering af et stort antal proteiner i identificerede celler/væv ved udvikling af X. laevis-embryoner. Tilgangen bygger på nøjagtig celleidentifikation, reproducerbare celleskæbnekort og etablerede metoder til at spore cellelinjer i denne biologiske model 6,7,8. Som vist i figur 1 studerer vi proteomer fra enkeltceller ved at anvende helcelledissektion eller kapillær mikrosampling til at aspirere cellulært indhold. Overvågning af en celles afstamning giver os mulighed for at studere proteomets rumlige temporale udvikling, når celler danner væv under gastrulation. Celleafkom markeres fluorescerende ved at injicere en fluorofor konjugeret til inert dextran eller mRNA til fluorescerende protein (fx grønt fluorescerende protein eller GFP). Det mærkede afkom isoleres på ønskede udviklingstidspunkter. Under gastrulation kan cellekloner, der er tæt grupperet, isoleres ved dissektion. Efter gastrulation kan cellekloner fordeles i embryoet på grund af migrationsbevægelser og kan isoleres fra dissocieret væv ved fluorescensaktiveret cellesortering (FACS). Proteiner i disse celler og væv måles via bottom-up proteomics ved hjælp af HPLC eller CE til separation og ESI tandem HRMS til identifikation. Celleafstamningsstyret HRMS-proteomics er skalerbar til forskellige cellestørrelser og slægter i embryoet og er specifik, følsom og kvantitativ. Gennem udvalgte eksempler vist her demonstrerer vi også, at denne protokol er skalerbar og bredt tilpasningsdygtig til forskellige typer celler og cellelinjer.
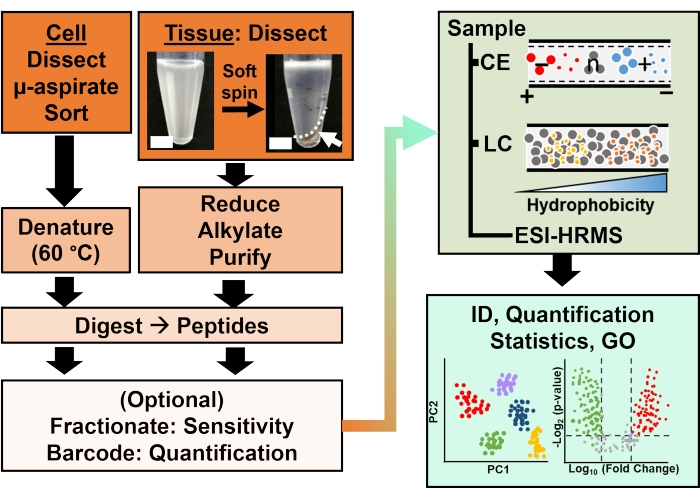
Figur 2: Den bioanalytiske arbejdsgang. Mikrodissektion og kapillær aspiration eller FACS lettede prøveudtagning af cellulært og klonalt proteinindhold. Udtømning af rigelige æggeblommeproteiner og adskillelse ved kapillær elektroforese (CE) eller nano-flow væskekromatografi (LC) forbedret identifikation (ID) følsomhed ved hjælp af elektrospray ionisering (ESI) massespektrometri med høj opløsning (HRMS). Kvantificering afslørede dysregulering og leverede ny information til hypotesedrevne undersøgelser i forbindelse med information tilgængelig fra genontologi (GO). Tallene er tilpasset med tilladelse fra reference15. Klik her for at se en større version af denne figur.
