Nuestra comprensión de la diferenciación celular y la génesis de tejidos y órganos es el resultado de décadas de elaboradas pantallas específicas de genes y sus productos. Aumentar nuestro conocimiento de todas las biomoléculas y sus cantidades durante eventos celulares importantes ayudaría a desentrañar los mecanismos moleculares que controlan el patrón espacial y temporal del plan corporal de los vertebrados. Las tecnologías que permiten la amplificación molecular y la secuenciación ahora pueden informar rutinariamente sobre un gran número de genes y transcripciones, apoyando estudios basados en hipótesis en investigación biológica básica y traslacional. Para comprender los sistemas en desarrollo, una relación compleja entre la transcripción y la traducción aboga por el análisis directo de múltiples proteínas y sus modificaciones postraduccionales. La proteómica global utilizando sistemas biológicos in vitro, como células madre pluripotentes inducidas, comenzó a delinear mecanismos de inducción tisular 1,2. En organismos complejos, como el embrión vertebrado, el desarrollo se basa en gradientes morfógenos en el contexto del espacio y el tiempo3. De ello se deduce que obtener conocimiento de los cambios proteómicos a medida que las células se diferencian para formar tejidos especializados, como los tejidos neurales, ofrece una clave para desbloquear programas moleculares que controlan el desarrollo normal y defectuoso y guiar la terapéutica de próxima generación.
La rana vertebrada sudafricana (Xenopus laevis) es un modelo bien establecido en biología celular y del desarrollo, neuro y regenerativa. El Premio Nobel de Fisiología o Medicina 2012 de Sir John Gurdon por el descubrimiento de la pluripotencia del núcleo somático destacó la importancia de este modelo para los descubrimientos en estudios básicos y traslacionales. Los embriones de Xenopus se desarrollan externamente a la madre, facilitando así la manipulación directa de células, clones celulares y expresión génica en varias etapas de desarrollo. La pigmentación asimétrica y las divisiones celulares estereotipadas permitieron el trazado de mapas de destino reproducibles del embrión de 7,8 células de 16-6 y 32 células. Para la proteómica basada en espectrometría de masas de alta resolución (HRMS), las ventajas adicionales del modelo incluyen un tamaño relativamente grande (~ 1 mm de diámetro), que produce un contenido abundante de proteínas para el análisis (~ 130 μg en embriones en etapa de escisión temprana, ~ 10 μg de contenido de proteína en células individuales del embrión de 16 células)9,10.
En la actualidad, HRMS es la tecnología líder de elección para detectar proteínas. Esta tecnología permite la detección y cuantificación directa, sensible y específica de múltiples, generalmente cientos a miles de proteínas diferentes11. La proteómica ascendente de HRMS implica una serie de pasos interconectados. Después de la extracción de la muestra de célula/tejido, las proteínas se digieren con una enzima proteolítica, como la tripsina (proteómica ascendente). Los péptidos resultantes se separan en función de sus diferentes propiedades fisicoquímicas, incluida la hidrofobicidad (cromatografía líquida de fase inversa, LC), la carga neta (cromatografía de intercambio iónico), el tamaño (cromatografía de exclusión de tamaño) o la movilidad electroforética (electroforesis capilar, CE). Los péptidos se cargan (ionizan), típicamente usando ionización por electrospray (ESI), y los iones peptídicos se detectan y secuencian a través de la fragmentación en fase gaseosa por HRMS en tándem. Los datos peptídicos resultantes se asignan al proteoma del organismo que se está estudiando. Con la intensidad de la señal de iones peptídicos específicos de proteínas (proteotípicos) que se correlacionan con la concentración, la cuantificación de proteínas se puede realizar sin etiqueta o basada en etiquetas (cuantificación de multiplexación). La proteómica HRMS proporciona un rico recurso de información sobre el estado molecular del sistema en estudio, lo que permite la generación de hipótesis y estudios funcionales de seguimiento.
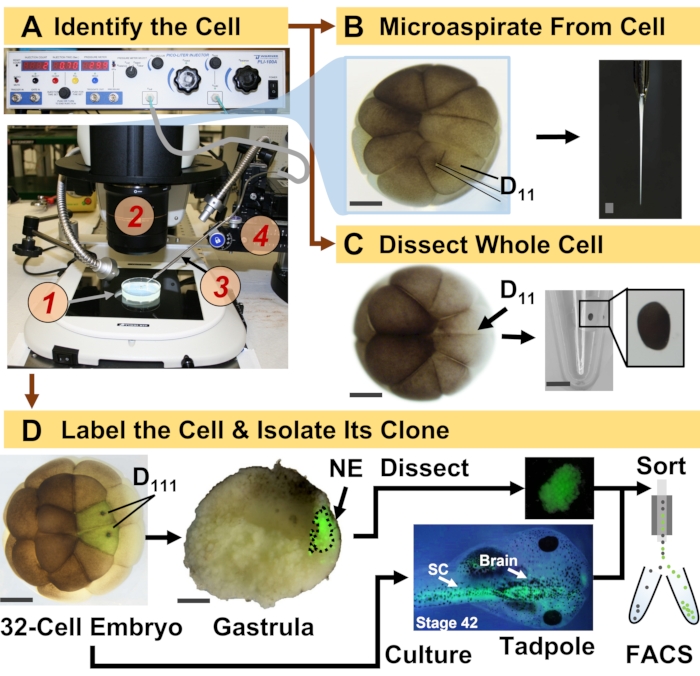
Figura 1: Proteómica escalable espaciotemporalmente que permite la proteómica HRMS guiada por linaje celular en el embrión en desarrollo (rana). (A) Visualización de la muestra (1) utilizando un microscopio estereoscópico (2) para la inyección de una célula identificada (recuadro), utilizando una micropipeta fabricada (3) bajo control por una etapa de traslación (4). (B) Muestreo subcelular de la célula D11 izquierda identificada en un embrión de 16 células. (C) Disección de una célula D11 completa de un embrión de 16 células. (D) Trazado fluorescente (verde) de las progenies D111 izquierda y derecha de un embrión de 32 células para guiar la disección del ectodermo neural (NE) en la gástrula (etapa 10) y el aislamiento del tejido descendente del renacuajo utilizando FACS. Barras de escala: 200 μm para embriones, 1,25 mm para el vial. Las figuras fueron adaptadas con permiso de las referencias 15,19,21,59. Haga clic aquí para ver una versión más grande de esta figura.
El protocolo presentado aquí permite la cuantificación basada en HRMS de un gran número de proteínas en células / tejidos identificados en embriones de X. laevis en desarrollo. El enfoque se basa en la identificación precisa de células, mapas de destino celular reproducibles y metodologías establecidas para rastrear linajes celulares en este modelo biológico 6,7,8. Como se muestra en la Figura 1, estudiamos proteomas de células individuales empleando disección de células enteras o micromuestreo capilar para aspirar el contenido celular. El monitoreo del linaje de una célula nos permite estudiar la evolución espacio-temporal del proteoma a medida que las células forman tejidos durante la gastrulación. La progenie celular se marca fluorescentemente inyectando un fluoróforo conjugado con dextrano inerte o ARNm para la proteína fluorescente (por ejemplo, proteína fluorescente verde o GFP). La progenie etiquetada se aísla en los puntos de tiempo de desarrollo deseados. Durante la gastrulación, los clones celulares que están estrechamente agrupados pueden aislarse por disección. Después de la gastrulación, los clones celulares pueden distribuirse dentro del embrión debido a movimientos migratorios y pueden aislarse de tejidos disociados mediante clasificación celular activada por fluorescencia (FACS). Las proteínas en estas células y tejidos se miden a través de la proteómica ascendente empleando HPLC o CE para la separación y ESI tándem HRMS para la identificación. La proteómica HRMS guiada por linaje celular es escalable a diferentes tamaños y linajes celulares dentro del embrión y es específica, sensible y cuantitativa. A través de ejemplos seleccionados que se muestran aquí, también demostramos que este protocolo es escalable y ampliamente adaptable a diferentes tipos de células y linajes celulares.
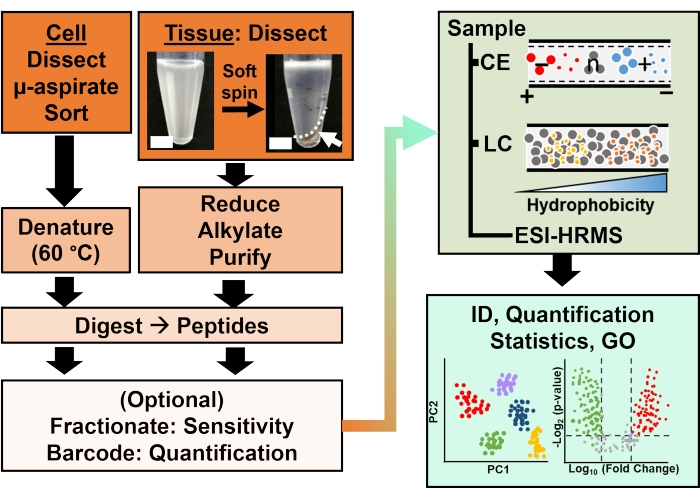
Figura 2: El flujo de trabajo bioanalítico. La microdisección y la aspiración capilar, o FACS, facilitaron el muestreo del contenido de proteínas celulares y clonales. Agotamiento de abundantes proteínas vitelinas y separación por electroforesis capilar (CE) o cromatografía líquida de nanoflujo (LC) mejora la sensibilidad de identificación (ID) utilizando espectrometría de masas de alta resolución (HRMS) por ionización por electrospray (ESI). La cuantificación reveló desregulación, proporcionando nueva información para estudios basados en hipótesis junto con la información disponible de la ontología génica (GO). Las figuras fueron adaptadas con permiso de la referencia15. Haga clic aquí para ver una versión más grande de esta figura.
