Unser Verständnis der Zelldifferenzierung und der Genese von Geweben und Organen ist das Ergebnis jahrzehntelanger aufwendiger gezielter Screenings von Genen und ihren Produkten. Ein besseres Wissen über alle Biomoleküle und ihre Mengen während wichtiger zellulärer Ereignisse würde dazu beitragen, molekulare Mechanismen zu entschlüsseln, die die räumliche und zeitliche Musterung des Körperbauplans von Wirbeltieren steuern. Technologien, die molekulare Amplifikation und Sequenzierung ermöglichen, sind nun in der Lage, routinemäßig über eine große Anzahl von Genen und Transkripten zu berichten und hypothesengestützte Studien in der biologischen und translationalen Grundlagenforschung zu unterstützen. Um sich entwickelnde Systeme zu verstehen, spricht eine komplexe Beziehung zwischen Transkription und Translation für die direkte Analyse mehrerer Proteine und ihrer posttranslationalen Modifikationen. Die globale Proteomik unter Verwendung biologischer In-vitro-Systeme, wie z. B. induzierter pluripotenter Stammzellen, begann, die Mechanismen der Gewebeinduktion zu beschreiben 1,2. In komplexen Organismen, wie dem Wirbeltierembryo, beruht die Entwicklung auf Morphogengradienten im Kontext von Raum und Zeit3. Daraus folgt, dass das Wissen über proteomische Veränderungen bei der Differenzierung von Zellen zu spezialisierten Geweben, wie z. B. Nervengeweben, einen Schlüssel zur Entschlüsselung molekularer Programme bietet, die die normale und fehlerhafte Entwicklung steuern, und um Therapeutika der nächsten Generation zu steuern.
Der südafrikanische Krallenfrosch (Xenopus laevis) ist ein etabliertes Modell in der Zell- und Entwicklungs-, Neuro- und regenerativen Biologie. Der Nobelpreisfür Physiologie oder Medizin 2012 von Sir John Gurdon 4,5 für die Entdeckung der Pluripotenz des somatischen Kerns unterstrich die Bedeutung dieses Modells für Entdeckungen in Grundlagen- und translationalen Studien. Xenopus-Embryonen entwickeln sich außerhalb der Mutter, was die direkte Manipulation von Zellen, Zellklonen und Genexpression über verschiedene Entwicklungsstadien hinweg ermöglicht. Asymmetrische Pigmentierung und stereotype Zellteilungen ermöglichten die Kartierung reproduzierbarer Schicksalskarten des 16-6- und 32-zelligen Embryos im 7,8-Stadium. Für die hochauflösende Massenspektrometrie (HRMS)-basierte Proteomik besteht ein weiterer Vorteil des Modells aus der relativ großen Größe (~1 mm Durchmesser), die einen hohen Proteingehalt für die Analyse liefert (~130 μg in Embryonen im frühen Spaltungsstadium, ~10 μg Proteingehalt in einzelnen Zellen des 16-zelligen Embryos)9,10.
Derzeit ist HRMS die führende Technologie der Wahl für den Nachweis von Proteinen. Diese Technologie ermöglicht den direkten, sensitiven und spezifischen Nachweis und die Quantifizierung von mehreren, in der Regel Hunderten bis Tausenden verschiedener Proteine11. Die Bottom-up-Proteomik von HRMS umfasst eine Reihe miteinander verbundener Schritte. Nach der Extraktion aus der Zell-/Gewebeprobe werden Proteine mit einem proteolytischen Enzym, wie z.B. Trypsin, verdaut (Bottom-up-Proteomik). Die resultierenden Peptide werden anhand ihrer unterschiedlichen physikalisch-chemischen Eigenschaften getrennt, einschließlich Hydrophobie (Umkehrphasen-Flüssigkeitschromatographie, LC), Nettoladung (Ionenaustauschchromatographie), Größe (Größenausschlusschromatographie) oder elektrophoretische Mobilität (Kapillarelektrophorese, CE). Die Peptide werden dann aufgeladen (ionisiert), typischerweise durch Elektrospray-Ionisation (ESI), und Peptidionen werden durch Gasphasenfragmentierung mittels Tandem-HRMS detektiert und sequenziert. Die resultierenden Peptiddaten werden auf das Proteom des untersuchten Organismus abgebildet. Mit proteinspezifischer (proteotypischer) Peptidionen-Signalintensität, die mit der Konzentration korreliert, kann die Proteinquantifizierung markierungsfrei oder markierungsbasiert (Multiplexing-Quantifizierung) durchgeführt werden. Die HRMS-Proteomik liefert eine reichhaltige Informationsquelle über den molekularen Zustand des untersuchten Systems, die die Generierung von Hypothesen und anschließende funktionelle Studien ermöglicht.
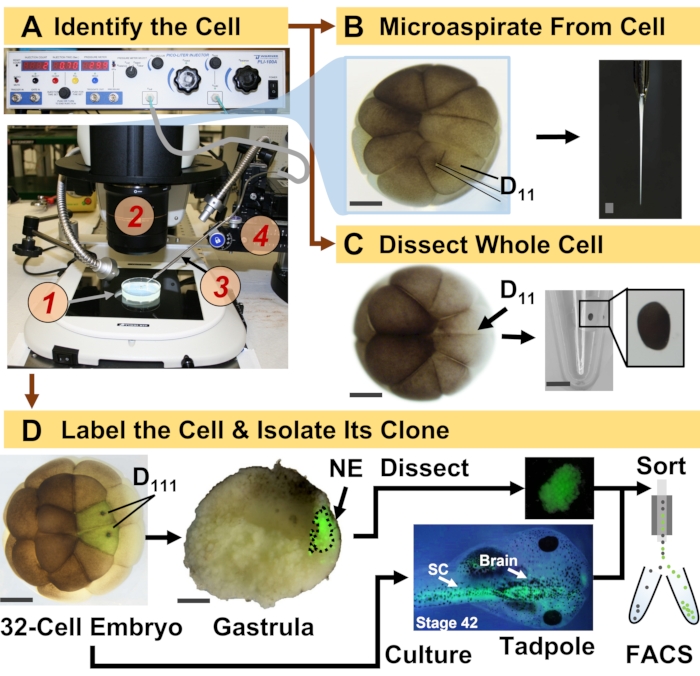
Abbildung 1: Räumlich-zeitlich skalierbare Proteomik, die eine zellliniengesteuerte HRMS-Proteomik im sich entwickelnden (Frosch-)Embryo ermöglicht. (A) Visualisierung der Probe (1) unter Verwendung eines Stereomikroskops (2) zur Injektion einer identifizierten Zelle (Inset) unter Verwendung einer hergestellten Mikropipette (3) unter Kontrolle durch einen Translationstisch (4). (B) Subzelluläre Probenahme der identifizierten linken D11-Zelle in einem 16-zelligen Embryo. (C) Präparation einer ganzenD-11-Zelle aus einem 16-zelligen Embryo. (D) Fluoreszierende (grüne) Nachverfolgung der linken und rechten D111-Nachkommen aus einem 32-zelligen Embryo zur Steuerung der Dissektion des neuralen Ektoderms (NE) in der Gastrula (Stadium 10) und Isolierung des Nachkommengewebes von der Kaulquappe mittels FACS. Maßstabsbalken: 200 μm für Embryonen, 1,25 mm für das Fläschchen. Die Abbildungen wurden mit Genehmigung der Referenzen 15,19,21,59 angepasst. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
Das hier vorgestellte Protokoll ermöglicht die HRMS-basierte Quantifizierung einer großen Anzahl von Proteinen in identifizierten Zellen/Geweben in sich entwickelnden X. laevis-Embryonen. Der Ansatz baut auf einer genauen Zellidentifikation, reproduzierbaren Zellschicksalskarten und etablierten Methoden zur Verfolgung von Zelllinien in diesem biologischen Modell auf 6,7,8. Wie in Abbildung 1 gezeigt, untersuchen wir Proteome aus einzelnen Zellen, indem wir Ganzzelldissektion oder Kapillarmikroskopie verwenden, um zellulären Inhalt abzusaugen. Die Überwachung der Abstammungslinie einer Zelle ermöglicht es uns, die raumzeitliche Entwicklung des Proteoms zu untersuchen, wenn Zellen während der Gastrulation Gewebe bilden. Die Nachkommen der Zelle werden fluoreszenzmarkiert, indem ein Fluorophor injiziert wird, der an inertes Dextran oder mRNA für fluoreszierendes Protein (z. B. grün fluoreszierendes Protein oder GFP) konjugiert ist. Die markierten Nachkommen werden zu den gewünschten Entwicklungszeitpunkten isoliert. Während der Gastrulation können Zellklone, die eng geclustert sind, durch Dissektion isoliert werden. Nach der Gastrulation können Zellklone aufgrund von Migrationsbewegungen innerhalb des Embryos verteilt und durch fluoreszenzaktivierte Zellsortierung (FACS) aus dissoziierten Geweben isoliert werden. Proteine in diesen Zellen und Geweben werden mittels Bottom-up-Proteomik unter Verwendung von HPLC oder CE zur Trennung und ESI-Tandem-HRMS zur Identifizierung gemessen. Die zellliniengesteuerte HRMS-Proteomik ist auf verschiedene Zellgrößen und Abstammungslinien innerhalb des Embryos skalierbar und spezifisch, sensitiv und quantitativ. Anhand ausgewählter Beispiele, die hier gezeigt werden, zeigen wir auch, dass dieses Protokoll skalierbar und breit an verschiedene Zelltypen und Zelllinien anpassbar ist.
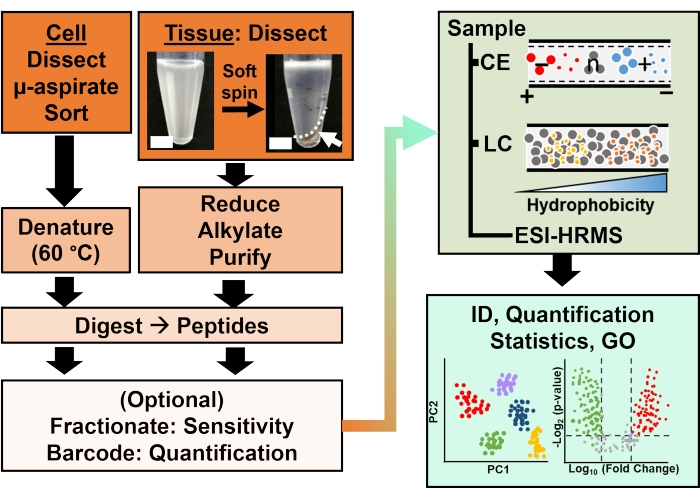
Abbildung 2: Der bioanalytische Arbeitsablauf. Die Mikrodissektion und Kapillaraspiration (FACS) erleichterte die Probenahme des zellulären und klonalen Proteingehalts. Die Depletion von reichlich vorhandenen Dotterproteinen und die Trennung durch Kapillarelektrophorese (CE) oder Nano-Flow-Flüssigkeitschromatographie (LC) verbesserte die Identifikationsempfindlichkeit (ID) unter Verwendung von hochauflösender Massenspektrometrie (HRMS) mit Elektrospray-Ionisation (ESI). Die Quantifizierung ergab eine Dysregulation, die neue Informationen für hypothesengetriebene Studien in Verbindung mit Informationen aus der Genontologie (GO) lieferte. Die Abbildungen wurden mit Genehmigung von Referenz15 angepasst. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
