Nossa compreensão da diferenciação celular e da gênese de tecidos e órgãos é o resultado de décadas de elaboradas telas direcionadas de genes e seus produtos. Aumentar nosso conhecimento de todas as biomoléculas e suas quantidades durante eventos celulares importantes ajudaria a desvendar mecanismos moleculares que controlam o padrão espacial e temporal do plano corporal dos vertebrados. Tecnologias que permitem amplificação molecular e sequenciamento são agora capazes de relatar rotineiramente um grande número de genes e transcritos, apoiando estudos orientados por hipóteses em pesquisa biológica básica e translacional. Para entender os sistemas em desenvolvimento, uma complexa relação entre transcrição e tradução defende a análise direta de múltiplas proteínas e suas modificações pós-traducionais. A proteômica global utilizando sistemas biológicos in vitro, como as células-tronco pluripotentes induzidas, começou a delinear mecanismos de indução tecidual 1,2. Em organismos complexos, como o embrião de vertebrados, o desenvolvimento depende de gradientes morfogênicos no contexto do espaço e do tempo3. Conclui-se que obter conhecimento das mudanças proteômicas à medida que as células se diferenciam para formar tecidos especializados, como tecidos neurais, oferece uma chave para desbloquear programas moleculares que controlam o desenvolvimento normal e defeituoso e orientar a terapêutica de próxima geração.
A rã-de-garra vertebrada sul-africana (Xenopus laevis) é um modelo bem estabelecido em biologia celular e de desenvolvimento, neuro e regenerativa. O Prêmio Nobel de Fisiologia ou Medicina de Sir John Gurdon em 2012 4,5 pela descoberta da pluripotência do núcleo somático destacou a importância desse modelo para descobertasem estudos básicos e translacionais. Os embriões de Xenopus desenvolvem-se externamente à mãe, facilitando assim a manipulação direta de células, clones celulares e expressão gênica ao longo de vários estágios de desenvolvimento. A pigmentação assimétrica e as divisões celulares estereotipadas permitiram o mapeamento de mapas reprodutíveis do destino do embrião de 16-6 e 32 célulasem 7,8 estádios. Para proteômica baseada em espectrometria de massa de alta resolução (EMAR), vantagens adicionais do modelo incluem tamanho relativamente grande (~1 mm de diâmetro), que produz conteúdo proteico abundante para análise (~130 μg em embriões em estágio inicial de clivagem, ~10 μg de conteúdo proteico em células isoladas do embrião de 16 células)9,10.
Atualmente, o HRMS é a tecnologia líder de escolha para detectar proteínas. Essa tecnologia permite a detecção e quantificação direta, sensível e específica de múltiplas, geralmente centenas a milhares de proteínas diferentes11. A proteômica ascendente por HRMS envolve uma série de etapas interconectadas. Após a extração da amostra de célula/tecido, as proteínas são digeridas com uma enzima proteolítica, como a tripsina (proteômica de baixo para cima). Os peptídeos resultantes são separados com base em suas diferentes propriedades físico-químicas, incluindo hidrofobicidade (cromatografia líquida de fase reversa, LC), carga líquida (cromatografia de troca iônica), tamanho (cromatografia de exclusão de tamanho) ou mobilidade eletroforética (eletroforese capilar, CE). Os peptídeos são então carregados (ionizados), tipicamente usando ionização por eletrospray (ESI), e íons peptídeos são detectados e sequenciados via fragmentação em fase gasosa por EMHR tandem. Os dados peptídicos resultantes são mapeados para o proteoma do organismo em estudo. Com a intensidade de sinal do peptídeo específico para proteínas (proteotípicas) correlacionando-se com a concentração, a quantificação da proteína pode ser realizada sem rótulos ou baseada em marcadores (quantificação multiplexação). A proteômica da HRMS fornece um rico recurso de informações sobre o estado molecular do sistema em estudo, permitindo a geração de hipóteses e estudos funcionais de acompanhamento.
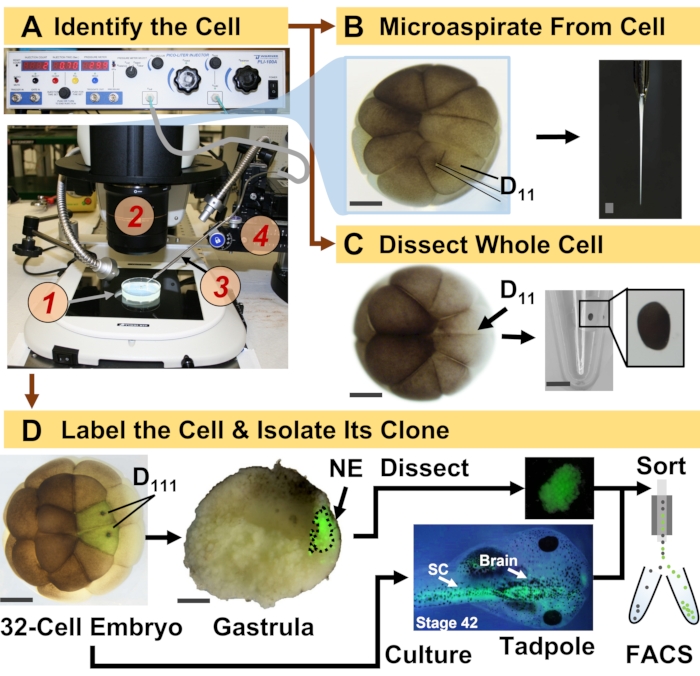
Figura 1: Proteômica espaço-temporalmente escalável permitindo a proteômica de HRMS guiada por linhagem celular no embrião em desenvolvimento (rã). (A) Visualização do espécime (1) utilizando um estereomicroscópio (2) para injeção de uma célula identificada (inset), utilizando uma micropipeta fabricada (3) sob controle por um estágio de translação (4). (B) Amostragem subcelular da célula D 11 esquerda identificada em um embrião de16 células. (C) Dissecção de uma célula D11 inteira de um embrião de 16 células. (D) Traçado fluorescente (verde) das progênies D111 esquerda e direita de um embrião de 32 células para guiar a dissecção do ectoderma neural (NE) na gástrula (estágio 10) e isolamento do tecido descendente do girino usando FACS. Barras de escala: 200 μm para embriões, 1,25 mm para o frasco para injetáveis. As figuras foram adaptadas com permissão das referências 15,19,21,59. Clique aqui para ver uma versão maior desta figura.
O protocolo aqui apresentado permite a quantificação baseada em HRMS de um grande número de proteínas em células/tecidos identificados em embriões de X. laevis em desenvolvimento. A abordagem baseia-se na identificação celular precisa, mapas de destino celular reprodutíveis e metodologias estabelecidas para rastrear linhagens celulares neste modelo biológico 6,7,8. Como mostrado na Figura 1, estudamos proteomas de células isoladas empregando dissecção de células inteiras ou microamostragem capilar para aspirar o conteúdo celular. O monitoramento da linhagem de uma célula nos permite estudar a evolução espaço-temporal do proteoma à medida que as células formam tecidos durante a gastrulação. A progênie celular é marcada fluorescentemente pela injeção de um fluoróforo conjugado a dextran inerte ou mRNA para proteína fluorescente (por exemplo, proteína fluorescente verde ou GFP). A progênie marcada é isolada nos momentos de desenvolvimento desejados. Durante a gastrulação, clones celulares que estão firmemente agrupados podem ser isolados por dissecção. Após a gastrulação, os clones celulares podem ser distribuídos dentro do embrião devido a movimentos migratórios e podem ser isolados de tecidos dissociados por triagem celular ativada por fluorescência (FACS). As proteínas nessas células e tecidos são medidas via proteômica bottom-up empregando HPLC ou CE para separação e ESI tandem HRMS para identificação. A proteômica da HRMS guiada por linhagens celulares é escalável para diferentes tamanhos e linhagens celulares dentro do embrião e é específica, sensível e quantitativa. Através de exemplos selecionados mostrados aqui, também demonstramos que este protocolo é escalável e amplamente adaptável a diferentes tipos de células e linhagens celulares.
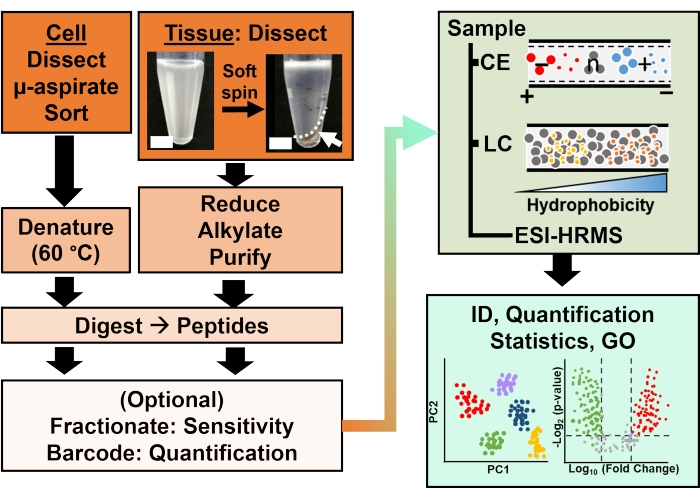
Figura 2: O fluxo de trabalho bioanalítico. A microdissecção e a aspiração capilar, ou FACS, facilitaram a amostragem do conteúdo de proteínas celulares e clonais. A depleção de proteínas vitelínicas abundantes e a separação por eletroforese capilar (CE) ou cromatografia líquida de nanofluxo (LC) aumentaram a sensibilidade de identificação (ID) usando espectrometria de massa de alta resolução (EMRH) com ionização por eletrospray (ESI). A quantificação revelou desregulação, fornecendo novas informações para estudos baseados em hipóteses em conjunto com informações disponíveis na ontologia gênica (GO). As figuras foram adaptadas com permissão da referência15. Clique aqui para ver uma versão maior desta figura.
