Vår förståelse av celldifferentiering och uppkomsten av vävnader och organ är resultatet av årtionden av utarbetade riktade skärmar av gener och deras produkter. Att öka vår kunskap om alla biomolekyler och deras kvantiteter under viktiga cellulära händelser skulle hjälpa till att avslöja molekylära mekanismer som styr det rumsliga och tidsmässiga mönstret för ryggradsdjurens kroppsplan. Teknologier som möjliggör molekylär amplifiering och sekvensering kan nu rutinmässigt rapportera om ett stort antal gener och transkript, vilket stöder hypotesdrivna studier inom grundläggande biologisk och translationell forskning. För att förstå utvecklingssystem förespråkar ett komplext förhållande mellan transkription och translation direkt analys av flera proteiner och deras posttranslationella modifieringar. Global proteomik som använder in vitro biologiska system, såsom inducerade pluripotenta stamceller, började avgränsa mekanismer för vävnadsinduktion 1,2. I komplexa organismer, såsom ryggradsdjurens embryo, är utvecklingen beroende av morfogengradienter i samband med rum och tid3. Det följer att få kunskap om proteomiska förändringar när celler differentieras för att bilda specialiserade vävnader, såsom neurala vävnader, erbjuder en nyckel för att låsa upp molekylära program som styr normal och defekt utveckling och vägleda nästa generations terapi.
Den sydafrikanska klogrodan (Xenopus laevis) är en väletablerad modell inom cell- och utvecklings-, neuro- och regenerativ biologi. Sir John Gurdons Nobelpris i fysiologi eller medicin 2012 4,5 för upptäckten av pluripotens hos den somatiska kärnan belyste vikten av denna modell för upptäckter i grundläggande och translationella studier. Xenopus-embryon utvecklas externt till modern, vilket underlättar direkt manipulation av celler, cellkloner och genuttryck över olika utvecklingsstadier. Asymmetrisk pigmentering och stereotypa celldelningar möjliggjorde kartläggning av reproducerbara ödeskartor från 16-6 och 32-celliga 7,8-stegs embryo. För högupplöst masspektrometri (HRMS) baserad proteomik inkluderar ytterligare fördelar med modellen relativt stor storlek (~ 1 mm i diameter), vilket ger rikligt proteininnehåll för analys (~ 130 μg i embryon i tidigt klyvningsstadium, ~ 10 μg proteininnehåll i enskilda celler i 16-cellembryot)9,10.
För närvarande är HRMS den ledande tekniken för att detektera proteiner. Denna teknik möjliggör direkt, känslig och specifik detektion och kvantifiering av flera, vanligtvis hundratusentals olika proteiner11. Bottom-up-proteomik av HRMS innebär en serie sammankopplade steg. Efter extraktion från cell/vävnadsprovet spjälkas proteiner med ett proteolytiskt enzym, såsom trypsin (bottom-up proteomik). De resulterande peptiderna separeras baserat på deras olika fysikalisk-kemiska egenskaper, inklusive hydrofobicitet (vätskekromatografi med omvänd fas, LC), nettoladdning (jonbyteskromatografi), storlek (storleksuteslutningskromatografi) eller elektroforetisk rörlighet (kapillärelektrofores, CE). Peptider laddas sedan (joniseras), vanligtvis med användning av elektrosprayjonisering (ESI), och peptidjoner detekteras och sekvenseras via gasfasfragmentering genom tandem HRMS. De resulterande peptiddata mappas till proteomet hos organismen som studeras. Med proteinspecifik (proteotypisk) peptidjonsignalintensitet korrelerad med koncentration kan proteinkvantifiering utföras etikettfri eller etikettbaserad (multiplexeringskvantifiering). HRMS-proteomik ger en rik resurs av information om det molekylära tillståndet i systemet som studeras, vilket möjliggör generering av hypoteser och uppföljande funktionella studier.
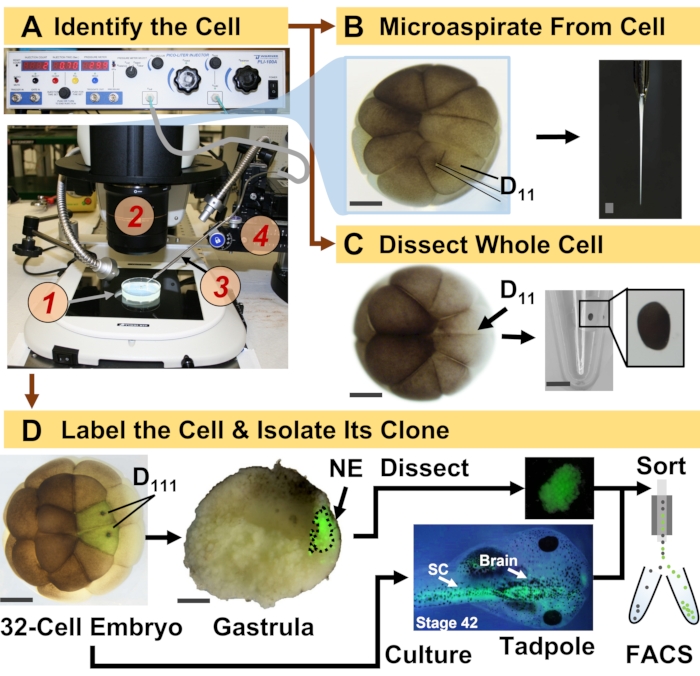
Figur 1: Spatiotemporalt skalbar proteomik som möjliggör cellinjestyrd HRMS-proteomik i det utvecklande (grod) embryot. (A) Visualisering av provet (1) med hjälp av ett stereomikroskop (2) för injektion av en identifierad cell (infälld) med hjälp av en tillverkad mikropipett (3) under kontroll genom ett translationssteg (4). b) Subcellulär provtagning av den identifierade vänstra D11-cellen i ett 16-celligt embryo. c) Dissektion av en hel D11-cell från ett embryo med 16 celler. (D) Fluorescerande (grön) spårning av vänster och höger D111-avkomma från ett 32-celligt embryo för att styra dissektion av neural ektoderm (NE) i gastrula (steg 10) och isolering av den nedstigande vävnaden från grodyngel med hjälp av FACS. Skalstänger: 200 μm för embryon, 1,25 mm för injektionsflaskan. Siffrorna anpassades med tillstånd från referenserna 15,19,21,59. Klicka här för att se en större version av denna figur.
Protokollet som presenteras här möjliggör HRMS-baserad kvantifiering av ett stort antal proteiner i identifierade celler/vävnader i utvecklande X. laevis embryon. Tillvägagångssättet bygger på noggrann cellidentifiering, reproducerbara cellödeskartor och etablerade metoder för att spåra cellinjer i denna biologiska modell 6,7,8. Som visas i figur 1 studerar vi proteom från enskilda celler genom att använda helcellsdissektion eller kapillär mikroprovtagning för att aspirera cellulärt innehåll. Övervakning av en cells härstamning gör det möjligt för oss att studera proteomets spatiotemporala utveckling när celler bildar vävnader under gastrulation. Cellavkomman markeras fluorescerande genom att injicera en fluorofor konjugerad till inert dextran eller mRNA för fluorescerande protein (t.ex. grönt fluorescerande protein eller GFP). Den märkta avkomman isoleras vid önskade utvecklingstidpunkter. Under gastrulation kan cellkloner som är tätt grupperade isoleras genom dissektion. Efter gastrulation kan cellkloner distribueras inom embryot på grund av migrationsrörelser och kan isoleras från dissocierade vävnader genom fluorescensaktiverad cellsortering (FACS). Proteiner i dessa celler och vävnader mäts via bottom-up-proteomik som använder HPLC eller CE för separation och ESI-tandem-HRMS för identifiering. Celllinjestyrd HRMS-proteomik är skalbar till olika cellstorlekar och linjer inom embryot och är specifik, känslig och kvantitativ. Genom utvalda exempel som visas här visar vi också att detta protokoll är skalbart och i stort sett anpassningsbart till olika typer av celler och cellinjer.
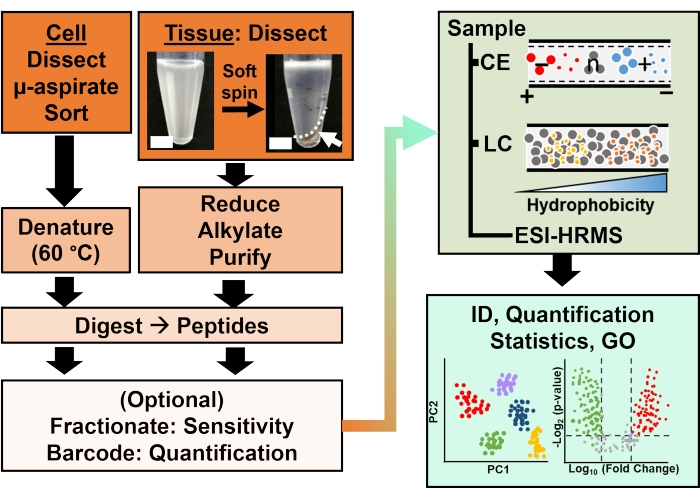
Figur 2: Det bioanalytiska arbetsflödet. Mikrodissektion och kapilläraspiration, eller FACS-underlättad provtagning av cellulärt och klonalt proteininnehåll. Utarmning av rikliga äggula proteiner och separation genom kapillärelektrofores (CE) eller nano-flöde vätskekromatografi (LC), förbättrad identifiering (ID) känslighet med hjälp av elektrospray jonisering (ESI) högupplöst masspektrometri (HRMS). Kvantifiering avslöjade dysregulering, vilket gav ny information för hypotesdrivna studier i kombination med information tillgänglig från genontologi (GO). Siffrorna har anpassats med tillstånd från referens15. Klicka här för att se en större version av denna figur.
