במהלך שני העשורים האחרונים, ביותרפיות התפתחו והפכו לעמוד התווך של תעשיית התרופות המודרנית. מגיפת SARS-CoV2 ומצבים מסכני חיים אחרים הגבירו עוד יותר את הצורך בפיתוח מהיר ורחב יותר של מולקולות ביו-פרמצבטיות 1,2,3.
המשקל המולקולרי הביותרפי הוא קריטי לזיהוי המולקולה, בשילוב עם בדיקות אנליטיות אחרות. מסות תת-היחידות השלמות והמופחתות משמשות לאורך מחזורי חיי הגילוי והפיתוח כחלק מאסטרטגיות בקרה שמטרתן לשמור על האיכות, כמתואר ב-QTPP (Quality Target Product Profile)4.
הפיתוח האנליטי בתעשיית הביו-פרמצבטיקה מסתמך במידה רבה על מדידות מסה לניתוח מסה שלם ואפיון עמוק באמצעות מיפוי פפטידים או ניטור בשיטה מרובת תכונות (MAM). במרכז טכניקות אלה המשתמשות בפלטפורמות ספקטרומטריית מסות מודרניות (MS) נמצאת היכולת לספק מדידות מסה מדויקות ברזולוציה גבוהה (HR/AM). רוב מכשירי HR/AM מניבים דיוקי מסה בטווח של 0.5-5 ppm, אשר מתרחבים עם טווח המסה. היכולת למדוד מסות במדויק עבור מולקולות גדולות שלמות מאפשרת זיהוי מהיר ובטוח של טיפולים במולקולות גדולות. מכיוון שלא ניתן להגיע לרזולוציה איזוטופית בתנאי ניסוי אופייניים למולקולות גדולות (>10 kDa), יש לחשב מסות ממוצעות לצורך השוואה וזיהוי 5,6.
ספקטרום מסה טיפוסי של חלבונים שלמים או תת-יחידות מייצג את הפרופיל הפרוטיאופורמי הכולל, המכיל מידע מרוכב על הצורות המולקולריות השונות הנובעות משינויים שלאחר התרגום (PTM) וכל הבדלי מבנה ראשוניים, כגון קליפים או גרסאות רצף. האופי הקל יחסית ובתפוקה הגבוהה של מדידות אלה הופך אותן לאטרקטיביות לאפיון וכבקרות ניטור בתהליך 7,8. ניתוח נתונים עבור ניסויים אלה דורש בדרך כלל מהמשתמש להגדיר את מרחב החיפוש עבור צורות מולקולריות (טווח של PTM או צורות מולקולריות אחרות). עבור חלבונים מסוכררים, מרחב חיפוש זה מונע במידה רבה על ידי הטרוגניות גליקוזילית. שילובים של PTM מרובים, תצורות קשר דיסולפידיות ווריאציות אחרות לאורך המבנה הראשוני הופכים את חישוב כל הצורות המולקולריות האפשריות למשימה מייגעת. לכן, החישוב הידני של הצורות המולקולריות האפשריות הוא תהליך גוזל זמן ומשאבים עם פוטנציאל גבוה לטעויות אנוש.
כאן, אנו מציגים כלי חישוב מסה שפותח בהתחשב בתכונות החשובות ביותר של מולקולות ביותרפיות, כגון mAbs, bsAbs, ADCs וכו ‘. הכלי מאפשר שילוב קל של משתני מרחב חיפוש לחישוב עקבי של מסות והרכבי יסודות. אופיו המודולרי של כלי זה יאפשר להמשיך לפתח אותו וליישם אותו לחישוב מסה והתאמת מסה לשיטות אחרות.
מודול ממשק המשתמש הגרפי מאפשר למשתמש לציין את הקלט לחישוב המסה, כפי שמוצג באיור 1; באופן ספציפי, המשתמש מזין רצפי חומצות אמינו של אות אחת עבור שרשראות נוגדנים קלות וכבדות. שינויים נפוצים עבור מחזוריות N-terminal בשרשרת כבדה וגזיר ליזין C-terminal כלולים כתיבות סימון. יתר על כן, ניתן להוסיף / להחסיר את הנוסחה הכימית / הרכב היסוד משרשראות חלבונים אלה באמצעות תיבת הטקסט המתאימה Chem Mod . זה מאפשר למשתמש את הגמישות להוסיף הרכב יסודי הכולל שינויים מרובים לאחר התרגום או מטען מולקולה קטנה במקרה של ADC. מכיוון שרוב ה- mAbs הטיפוליים מתוכננים להסיר את אתרי הגליקוזילציה בשרשרת האור, גליקוזילציה בשרשרת האור נותרת אופציונלית וניתן להגדיר אותה באמצעות תיבת סימון בממשק המשתמש הגרפי.
וריאציה טיפוסית על ניתוח מסה שלמה עבור נוגדנים היא ניתוח מסה תת-יחידה מופחתת, שבו השרשרת הקלה מנותקת מהשרשרת הכבדה על ידי הפחתת הקשרים הדיסולפידים הבין-שרשרתיים. בהתאם לחוזק של סוכן מחזרים בשימוש, קשרים דיסולפיד intrachain עשוי או לא יכול להיות בקע. למשתמשים יש את הגמישות להזין את המספר הכולל של אג”ח דיסולפיד בהתאם לתת-סוג IgG או במקרה של ADC מצומד ציסטאין9.
היישום מחשב מסות באופן מלמטה למעלה, שבו ההרכבים היסודיים מחושבים תחילה עבור שרשראות כבדות בודדות ושרשראות קלות. לאחר מכן, שרשרת כבדה (HC) N-terminal cyclization Lys-clipping מטופל על ידי התאמת הרכבי היסוד מחושבים. כל שינוי כימי שצוין מוחל לאחר מכן על השרשראות הכבדות ו / או הקלות. בהתאם לסוג הניתוח ולדפוסי הקשר הדיסולפידים שצוינו על ידי המשתמש, מספר המימנים מותאם לשתי שרשראות הפוליפפטידים. המסות HC גליקוזילציה ושרשרת אור (LC) (אופציונליות) מחושבות בהתבסס על הקלט של המשתמש. לבסוף, מסות HC ו- LC מרובות משולבות, ומספרי הקשר הדיסולפידים מתעדכנים אוטומטית לחישוב המסה השלם.
עם מולקולות גדולות יותר כגון חלבונים שלמים, לא ניתן למדוד מסות מונואיזוטופיות בגלל פגם המסה התוסף בעת שימוש בספקטרומטרים של מסות עם כוח פתרון אופייני. במקום זאת, מסות נומינליות או ממוצעות נמדדות או מדווחות 5,10,11,12,13. מסות היסוד הממוצעות יכולות להשתנות בהתאם למקור המשמש למסות14,15. בעוד שההבדלים במסות היסוד עשויים להיות קטנים, הם יכולים להצטבר לערכים משמעותיים עבור חישובי משקל מולקולרי של מולקולות גדולות. המסות היסודיות הממוצעות המשמשות כברירת מחדל ביישום התוכנה מוצגות בטבלה משלימה 1. עבור סביבות מוסדרות כמו מחקר ופיתוח ביו-פרמצבטי (R&), חשוב לשמור על מסות מולקולריות עקביות מכיוון ששינויים במסות עשויים לרמוז על שינויים בישות המולקולרית במהלך הגשות רגולטוריות. כדי לאפשר עקביות בשימוש במסות יסוד, מילון של מסות יסוד נכלל בכלי התוכנה כקובץ טקסט של ערכים מופרדים באמצעות פסיקים (csv): Element_Mass.csv (קובץ קידוד משלים 1). באופן דומה, רשימה של הרכבי גליקן הנראים בדרך כלל ב- mAbs כלולה: גליקן.csv (קובץ קידוד משלים 2). שני הקבצים נשמרים באותו מיקום תיקיה כמו יישום הפעלה, והמשתמש יכול לשנות אותם כדי להשתמש ברשימת מסת רכיבים ספציפית או בספריית גליקן.
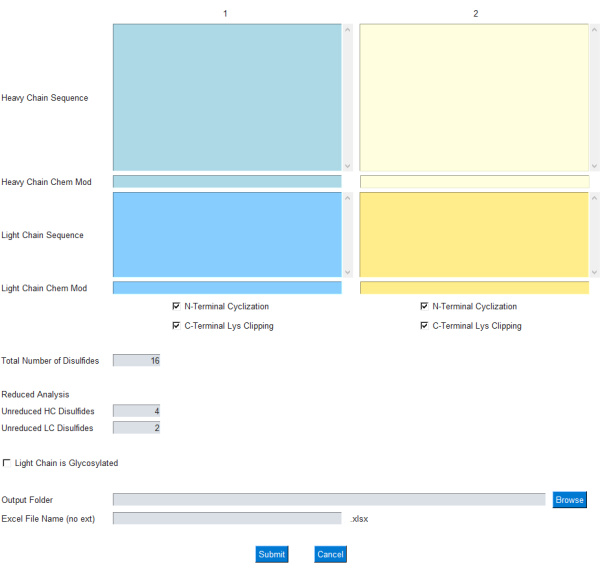
איור 1: ממשק GUI עבור יישום mAbScale. מודול GUI מאפשר למשתמש לציין את הקלט לחישוב המסה. המשתמש מזין רצפי חומצות אמינו בנות אות אחת עבור שרשראות הנוגדנים הקלות והכבדות. שינויים נפוצים עבור מחזוריות N-terminal בעלת שרשרת כבדה וגזיר ליזין מסוף C כלולים כתיבות סימון. נוסחאות כימיות / הרכבי יסודות ניתן להוסיף / להחסיר באמצעות תיבת הטקסט המתאימה Chem Mod . אנא לחץ כאן כדי להציג גרסה גדולה יותר של איור זה.
