지난 20년 동안 바이오 치료제는 현대 제약 산업의 중심이 되도록 발전했습니다. SARS-CoV2 팬데믹 및 기타 생명을 위협하는 조건으로 인해 바이오 제약 분자 1,2,3의 더 빠르고 광범위한 개발에 대한 필요성이 더욱 높아졌습니다.
바이오치료제 분자량은 다른 분석 분석법과 함께 분자를 식별하는 데 매우 중요합니다. 온전한 소단위 질량과 감소된 소단위 질량은 QTPP(Quality Target Product Profile)4에 설명된 대로 품질 유지를 목표로 하는 제어 전략의 일환으로 발견 및 개발 수명 주기 전반에 걸쳐 사용됩니다.
바이오 제약 산업의 분석 개발은 펩타이드 맵핑 또는 다중 속성 분석법(MAM) 모니터링을 사용한 원형(intact) 질량 분석 및 심층 특성 분석을 위한 질량 측정에 크게 의존합니다. 최신 질량분석법(MS) 플랫폼을 활용하는 이러한 기술의 핵심은 고분해능 정밀 질량(HR/AM) 측정을 제공하는 기능입니다. 대부분의 HR/AM 기기는 0.5-5ppm 범위의 질량 정확도를 제공하며, 이는 질량 범위에 따라 확장됩니다. 온전한 거대 분자에 대한 질량을 정확하게 측정할 수 있는 능력은 거대 분자 치료제를 빠르고 확실하게 식별할 수 있게 합니다. 큰 분자 (>10 kDa)에 대한 일반적인 실험 조건을 사용하여 동위원소 분해능을 얻을 수 없기 때문에 비교 및 식별을 위해 평균 질량을 계산해야합니다 5,6.
일반적인 원형(intact) 또는 소단위(subunit) 단백질 질량 스펙트럼은 전사 후 변형(post-translational modification, PTM) 및 클립 또는 염기서열 변이체(sequence variant)와 같은 1차 구조 차이로 인한 다양한 분자 형태에 대한 복합 정보를 포함하는 전체 단백질형 프로파일을 나타냅니다. 이러한 측정은 상대적으로 쉽고 처리량이 많기 때문에 특성화 및 공정 중 모니터링 제어에 적합합니다 7,8. 이러한 실험에 대한 데이터 분석은 일반적으로 사용자가 분자 형태(PTM 범위 또는 기타 분자 형태)에 대한 검색 공간을 정의해야 합니다. 당화 단백질의 경우, 이 검색 공간은 주로 당형(glycoform) 이질성에 의해 주도됩니다. 여러 PTM, 이황화물 결합 구성 및 기본 구조에 따른 기타 변형의 조합으로 인해 가능한 모든 분자 형태를 계산하는 것은 지루한 작업입니다. 따라서 가능한 분자 형태의 수동 계산은 인적 오류가 발생할 가능성이 높은 시간과 자원 소모적인 프로세스입니다.
여기에서는 mAb, bsAbs, ADC 등과 같은 바이오 치료제 분자의 가장 중요한 특징을 고려하여 개발된 질량 계산 도구를 소개합니다. 이 도구를 사용하면 질량 및 원소 조성의 일관된 계산을 위해 검색 공간 변수를 쉽게 통합할 수 있습니다. 이 도구의 모듈식 특성으로 인해 추가 개발이 가능하여 다른 양식에 대한 질량 계산 및 질량 매칭에 적용할 수 있습니다.
GUI 모듈을 통해 사용자는 그림 1과 같이 질량 계산을 위한 입력을 지정할 수 있습니다. 특히, 사용자는 가벼운 항체 사슬과 무거운 항체 사슬에 대한 단일 문자 아미노산 서열을 입력합니다. 중쇄 N-말단 고리화 및 C-말단 라이신 클리핑에 대한 일반적인 수정이 확인란으로 포함되어 있습니다. 또한, 화학식/원소 조성은 각각의 Chem Mod 텍스트 상자를 통해 이러한 단백질 사슬에서 추가/뺄 수 있습니다. 이를 통해 사용자는 여러 번역 후 변형 또는 ADC의 경우 저분자 페이로드를 포함하는 원소 조성을 유연하게 추가할 수 있습니다. 대부분의 치료용 mAb는 경쇄의 당화 부위를 제거하도록 설계되었기 때문에 경쇄의 당화는 선택 사항이며 GUI의 확인란을 사용하여 지정할 수 있습니다.
항체에 대한 원형 질량 분석의 일반적인 변형은 사슬 간 이황화 결합을 감소시켜 경쇄가 중쇄에서 분리되는 감소된 소단위 질량 분석입니다. 사용된 환원제의 강도에 따라 사슬 내 이황화물 결합이 절단되거나 절단되지 않을 수 있습니다. 사용자는 IgG 아형에 따라 또는 시스테인 복합 ADC9의 경우 이황화 결합의 총 수를 입력할 수 있는 유연성을 갖습니다.
이 응용 프로그램은 상향식 방식으로 질량을 계산하며, 여기서 원소 조성은 개별 중쇄 및 경쇄에 대해 먼저 계산됩니다. 다음으로, 중쇄(HC) N-말단 고리화 Lys-클리핑은 계산된 원소 조성을 조정하여 설명됩니다. 그런 다음 지정된 화학적 변형이 중쇄 및/또는 경쇄에 적용됩니다. 분석 유형 및 사용자가 지정한 이황화 결합 패턴에 따라 수소 수는 두 개의 폴리펩티드 사슬에 대해 조정됩니다. 당화 HC 및 경쇄(LC)(선택 사항) 질량은 사용자의 입력에 따라 계산됩니다. 마지막으로, 여러 HC 및 LC 질량이 결합되고 온전한 질량 계산을 위해 이황화물 결합 번호가 자동으로 업데이트됩니다.
원형(intact) 단백질과 같은 더 큰 분자의 경우, 일반적인 분해능을 가진 질량 분석기를 사용할 때 첨가 질량 결함으로 인해 단일 동위원소 질량을 측정할 수 없습니다. 대신, 공칭 또는 평균 질량이 측정되거나 보고됩니다 5,10,11,12,13. 평균 원소 질량은 선별된 질량14,15에 사용된 소스에 따라 달라질 수 있습니다. 원소 질량의 차이는 작을 수 있지만 대분자 분자량 계산에서 중요한 값을 합산할 수 있습니다. 소프트웨어 응용 프로그램에서 기본적으로 사용되는 평균 원소 질량은 보충 표 1에 나와 있습니다. 바이오 제약 연구 및 개발(R&D) 분야와 같은 규제 환경의 경우, 질량의 변화는 규제 서류 제출 중에 분자 실체의 변화를 의미할 수 있기 때문에 일관된 분자 질량을 유지하는 것이 중요합니다. 원소 질량 사용의 일관성을 유지하기 위해 원소 질량 사전이 소프트웨어 도구에 쉼표로 구분된 값(csv) 텍스트 파일인 Element_Mass.csv(보충 코딩 파일 1)로 포함되어 있습니다. 유사하게, mAb에서 일반적으로 볼 수 있는 글라이칸 조성물의 선별된 목록이 포함되어 있습니다: Glycan.csv(보충 코딩 파일 2). 두 파일 모두 실행 가능한 응용 프로그램과 동일한 폴더 위치에 저장되며 사용자가 특정 원소 질량 목록 또는 글라이칸 라이브러리를 사용하도록 수정할 수 있습니다.
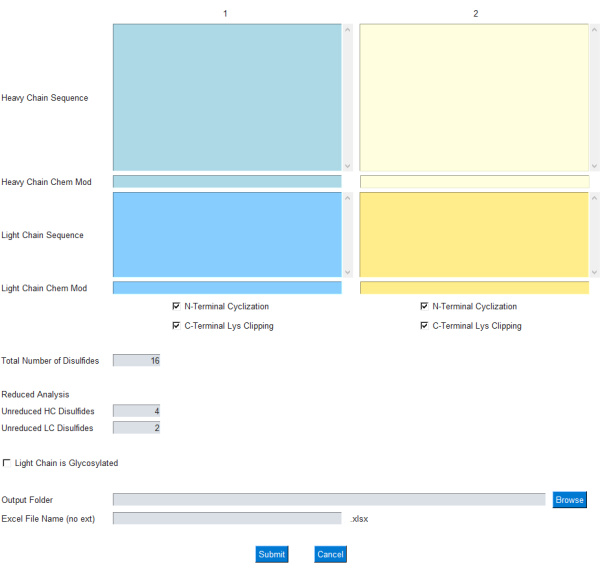
그림 1: mAbScale 응용 제품을 위한 GUI 인터페이스. GUI 모듈을 통해 사용자는 질량 계산을 위한 입력을 지정할 수 있습니다. 사용자는 가벼운 항체 사슬과 무거운 항체 사슬에 대한 단일 문자 아미노산 서열을 입력합니다. 중쇄 N-말단 순환화 및 C-말단 라이신 클리핑에 대한 일반적인 수정이 확인란으로 포함되어 있습니다. 화학식/원소 조성은 해당 Chem Mod 텍스트 상자를 통해 더하거나 뺄 수 있습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
