Figure 1 gives a schematic overview of the different methods described in this article. 1) Mice receive an injection of MOG35-55 antigen and develop initial clinical symptoms after 10.7 ± 0.3 days28. A representative disease course of EAE mice is shown in Figure 1. 2) Various tissues (spleen, lymph nodes, lumbar spinal cord) and blood are extracted from control and EAE mice at different time points during the preclinical phase (day 2, day 4, day 6), during the onset of the disease (day 10) and during the acute phase of the disease (day 12, day 14). 3) The mRNA expression profile of cytokines, which regulate EAE development, is determined in the various tissues extracted at different time points during the disease course, using quantitative PCR. 4) Single cells are isolated from various tissues extracted at different time points during the disease course and the immune cell distribution determined using flow cytometry.
In spleen and lymph nodes, T cells and B cells are observed as the most prominent cell type. In contrast to lymph nodes, in the spleen, additionally, a high number of monocytes and neutrophils are present. All immune cells show a transient increase in the lymph nodes during the acute phase, whereas only macrophages, B cells, T cells and neutrophils increase in the spleen. In blood, all immune cells increase transiently during the preclinical phase. In the lumbar spinal cord, a disease-dependent increase in all immune cells is observed, apart from monocytes, which presumably differentiate to macrophages after their entrance into the spinal cord (Figure 2). CD4+ and CD8+ T cells show comparable changes in spleen, lymph node and blood during the different disease courses. Among cells infiltrating the spinal cord, the increase in CD4+ T cells was most pronounced (Figure 3). In Figure 4, the gating-strategy for the determination of the different cell populations is shown. In detail, cell populations described in this manuscript were identified as follows: monocytes (CD45hi, CD3-, Ly6G-, CD19-, CD11c-, CD11b+), macrophages (CD45hi, CD3-, Ly6G-, CD11c-, CD11b+, F4/80hi), neutrophils (CD45hi, CD3-, CD11b+, Ly6G+), dendritic cells (CD45hi, CD3-, Ly6G-, CD19-, CD11c+), B cells (CD45hi, CD3-, CD19+), T cells (CD45hi, CD3+), CD4+ T cells (CD45hi, CD3+, CD4+) and CD8+ T cells (CD45hi, CD3+, CD8+). The numbers of cells are related to the amount of tissue (weight of spleen, lymph nodes, lumbar spinal cord) or the blood volume, thus reflecting the absolute cell count. It is, however, possible to express the cell types as percentages of total cells measured.
In lymph nodes, the expression of IL-23 mRNA is transiently increased during the preclinical phase, whereas the expression of IL-6 mRNA is increased in a disease-dependent manner. In the spleen, IL-6 and IL-23 mRNA expression increases transiently in the preclinical phase, while at the onset of the disease, IL-1β mRNA expression is raised. In blood, the expression of TNFα mRNA is increased in a disease-dependent manner. In the lumbar spinal cord, TNFα, IL-1β and IFNγ increase during the acute phase, whereas IL-6 is upregulated during the onset phase (Figure 5).
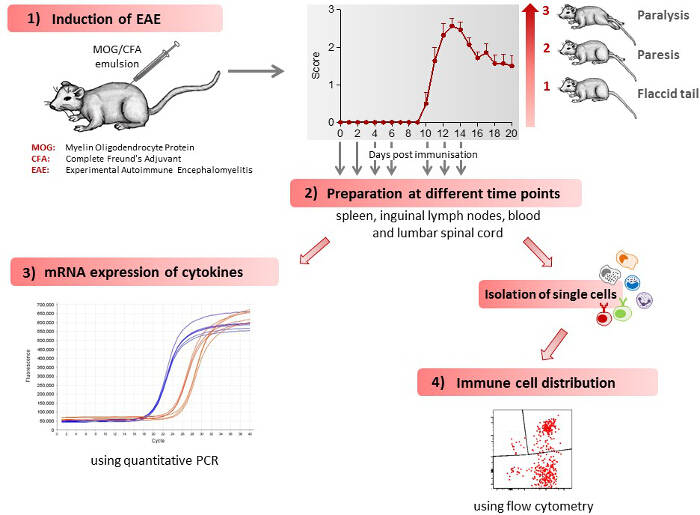
Figure 1: Scheme of the Work Flow for Induction and Immunological Assessment of EAE. 1) EAE is induced in mice leading to the development of clinical symptoms. Clinical symptoms are classified by clinical scores, as follows: 0) no signs, 0.5) distal paralysis of the tail, 1) complete paralysis of the tail, 1.5) limp tail and mild weakness of hind legs, 2) limp tail and severe weakness of hind legs, 2.5) limp tail and paralysis of one hind leg, 3) limp tail and paralysis of both hind legs. The number of mice used in the figure shown was 7. The figure has been modified from Barthelmes et al. 28. 2) The mice are sacrificed at different time points during the disease course, spleen, inguinal lymph nodes, blood and lumbar spinal cord extracted and single cells isolated from these tissues. 3) Immune cell distribution in various tissues, as determined by flow cytometry. 4) mRNA expression for various cytokines in the different tissues, as determined by quantitative PCR. Please click here to view a larger version of this figure.
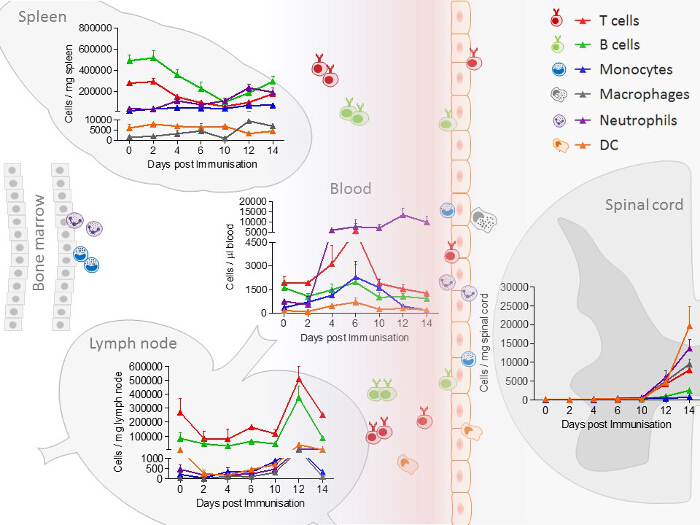
Figure 2: Immune Cell Distribution, Determined by FACS (Fluorescence-Activated Cell Sorting) Analysis in Spleen, Lymph node, Blood and Spinal Cord Samples from EAE Mice at Different Disease Stages. In the experiment shown, the number of mice per group was 2 – 10. The distribution of neutrophils/monocytes in blood and neutrophils/macrophages in the spinal cord are adapted and modified from Barthelmes et al.28. Please click here to view a larger version of this figure.
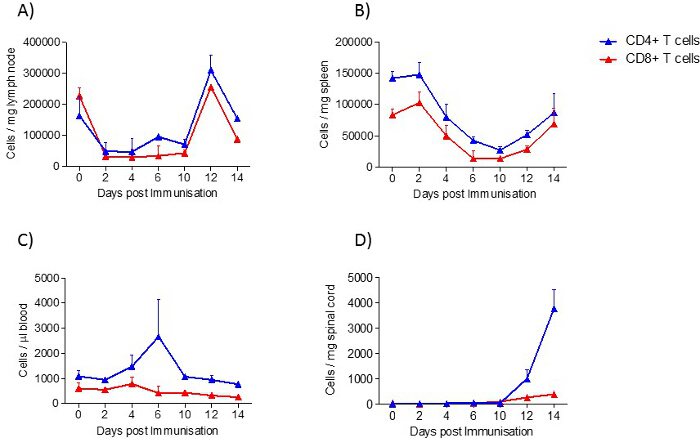
Figure 3: CD4+ T cell and CD8+ T cell Distribution in Samples from EAE Mice at Different Disease Stages, Determined by FACS Analysis. A) lymph node, B) spleen, C) blood and D) spinal cord. In the experiment shown, the number of mice per group was 2 – 10. Results are presented as means ± standard errors (SEM). Please click here to view a larger version of this figure.
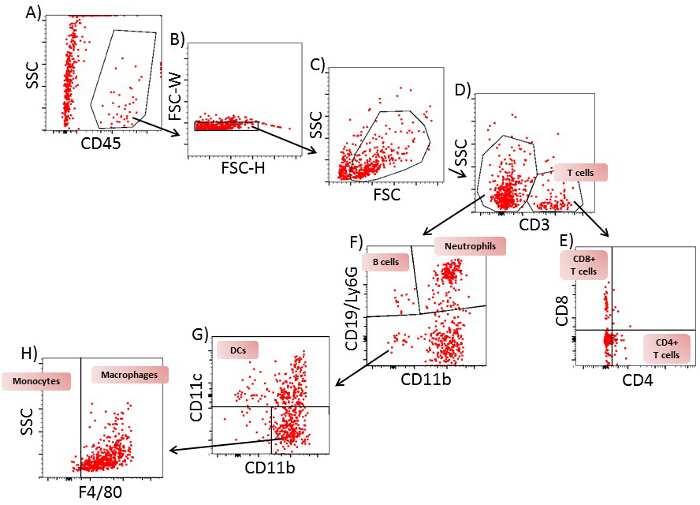
Figure 4: Gating Strategy for Flow Cytometric Analysis of T Cells, B Cells, Neutrophils, Monocytes, Dendritic Cells and Macrophages from Spinal Cord of EAE Mice on Day 12. A) A plot of SSC/CD45 from all cells. Gate: CD45+ cells. B) A plot of FSC-H/FSC-W from CD45+ cells. Gate: single cells. C) A plot of SSC-A/FSC-A from single cells. Gate: viable cells. D) A plot of FSC-H/CD3 from viable cells. Gate: CD3+ and CD3– cells. E) A plot of CD4/CD8 from CD3+ cells. Gate: CD4+CD8– (CD4+ T cells) and CD4–CD8+ (CD8+ T cells). F) A plot of CD19 and Ly6G/CD11b from CD3– cells. Gate: CD19+CD11b– cells (B cells), Ly6G+CD11b+ cells (neutrophils) and CD19–Ly6G– cells. G) A plot of CD11c/CD11b cells from CD19–Ly6G– cells: Gate CD11c+ cells (dendritic cells) and CD11c– cells. H) A plot of SSC/F4/80 from CD11c– cells. Gate: F4/80– cells (monocytes) and F4/80+ cells (macrophages). One representative plot of 9 is shown. Please click here to view a larger version of this figure.
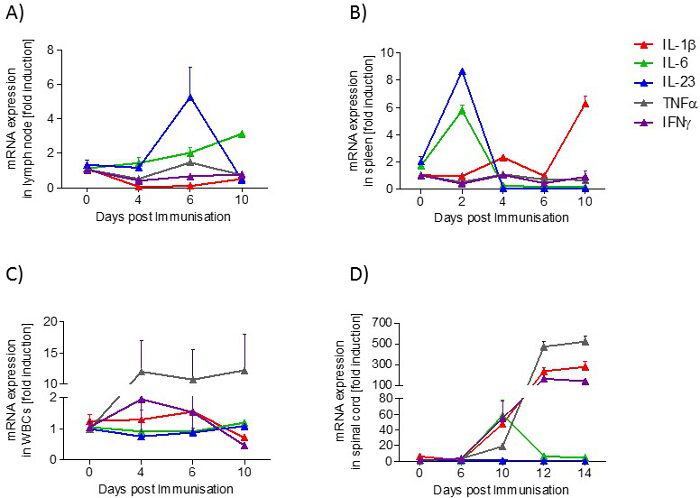
Figure 5: Cytokine Profile (IL-1β, IL-6, IL-23, TNFα, IFNγ) in Samples from EAE Mice at Different Disease Stages. A) lymph node, B) spleen, C) blood and D) spinal cord. The mRNA expression levels were normalized to peptidyl propyl isomerase A (PPIA) and were calculated using the mRNA level of untreated mice of the same age. Measurements were done in triplicate. The number of mice per group was 2 – 3. Results are presented as means ± standard errors (SEM). Please click here to view a larger version of this figure.
| Gene | forward | reverse | fragement size (Bp) | ||||
| IL-1β | CTGGTGTGTGACGTTCCCATTA | CCGACAGCACGAGGCTTT | 76 | ||||
| IL-6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC | 76 | ||||
| IL-23 | GAGCAGCAACTCTGACTGAGCC | GAACAGCACAAGTCCTAATGGGTTA | 127 | ||||
| IFNγ | CACGGCACAGTCATTGAAAGC | CACCATCCTTTTGCCAGTTCC | 118 | ||||
| TNFα | TGACAAGCCTGTAGCCCACG | GCCTTGTCCCTTGAAGAGAACC | 178 | ||||
| PPIA | GCTGGACCAAACACAAACGG | GCCATTCCTGGACCCAAAAC | 144 | ||||
Table 1: Primer pairs.
