In this study, we describe the application of the polysomal profiling technique to three different sources: parasitic Leishmania major, cultured human cells, and mouse testis. Leishmania cells freely grow in the liquid media in suspension, cultured human cells grow in the adherent monolayer on plates, and the mouse testis represents a tissue sample. The method can be easily adjusted to other types of freely grown cells in suspension, different types of tissues, or from another organism, and different types of cultured cells. The approach consists of four major steps: lysate preparation, a sucrose gradient preparation and ultracentrifugation step, polysome fractionation and sample collection followed by analysis of the fractions. Cells from different sources are collected, washed and lysed in lysis buffer by passage through a needle or Dounce homogenizer. Centrifugation is used to remove cell debris, clarifying the lysate. The scheme of gradient fractionation is shown in Figure 1A. A continuous sucrose gradient is formed by the mixing of 10% and 50% sucrose solutions in a gradient maker. The lysate is loaded on the top of the gradient. Ultracentrifugation separates mRNAs associated with a different number of ribosomes which is monitored by a UV detector during fractionation, forming a distinct absorbance spectrum. Collected fractions are used for RNA and protein analysis (Figure 1B). RNA can be analyzed by electrophoresis followed by Northern blot or used for cDNA production followed by a RT-qPCR reaction to analyze the association of individual mRNAs with polysomes. Next-generation sequencing can be used to analyze the translational status of mRNAs on a genome-wide scale7. For protein analysis of polysomal fractions, proteins are precipitated with trichloroacetic acid to concentrate them. Proteins are then analyzed by Western blotting or by mass spectroscopy at the proteome level.
A typical polysomal profile generated from Leishmania major actively growing culture is shown in Figure 2A. The absorbance graph of the fractionation has a distinct shape with typical peaks for ribosome subunits (40S and 60S), single ribosomes (80S or monosomes) and polysomes.
Quantitative RT-PCR (RT-qPCR) was employed to detect association of individual Leishmania mRNAs with ribosomes and polysomes. The comparative CT (ΔΔCT)31 method is a simple and appropriate approach to study relative mRNAs levels in the cells. This method requires an internal control (a stable mRNA, that does not change expression during treatments or conditions of the experiment) for calculations. However, there is no internal control in polysomal fractions because levels of any mRNA or ribosomal RNA will vary in fractions, depending on their association with ribosomes, polysomes, etc. To solve the problem of an internal control, we have used synthetic bacterial OmpA mRNA for normalization of relative individual Leishmania mRNA levels in the fractions. OmpA mRNA was synthesized in vitro and added in equal amounts to each fraction before RNA extractions. Addition of the synthetic RNA is important because it makes calculations of RT-qPCR data more precise, serving as internal control for calculations by comparative CT (ΔΔCT) method.
Aliquots of the gradient fractions were mixed into three groups: prepolysomes (subunits and monosomes), light polysomes (consisting of 2-4 ribosomes), and heavy polysomes (consisting of 5-8 ribosomes). RT-qPCR was performed on RNA from combined fractions to analyze mRNA distribution between these combined fractions (Figure 2B). 18S ribosomal RNA was used as a control. Its relative levels determined by RT-qPCR correlated well with the estimated distribution of the small ribosomal subunits (free subunit, and as a part of monosomes and polysomes) on the spectrum. RT-qPCR analysis revealed that individual mRNAs tested have a different degree of engagement in translation during logarithmic phase of Leishmania growth. Tubulin mRNA is associated preferentially with heavy polysomes, suggesting efficient translation. In contrast, Sherp mRNA is found primarily with prepolysomes and light polysomes supporting less active translation in comparison with tubulin mRNA.
Expression of recombinant proteins in cultured cells is an important experimental approach in diverse categories of studies. Here, we present an example of polysomal profiling of recombinant protein mRNAs in another sample source, cultured human cells. HeLa cells were transiently transfected with plasmids expressing recombinant cystic fibrosis transmembrane conductance regulator (CFTR)34 or Norrie disease protein (NDP). Absorbance spectra of polysome fractionation from these two independent cultures were very similar and contained distinct peaks corresponding ribosomal subunits (40S and 60S), monosomes (80S), and polysomes (Figure 3A). Similarity in the spectra from these experiments illustrates reproducibility of the gradient fractionation. Like in the Leishmania studies, the distribution of mRNAs was determined by RT-qPCR in the fractions representing prepolysomes, light polysomes, and heavy polysomes (Figure 3B). Detection of small ribosomal subunit 18S RNA correlated with their estimated distribution in the spectra. NDP mRNAs were mostly associated with light and heavy polysomal fractions, while CFTR mRNAs were mostly found in prepolysome fractions, suggesting that NDP is translated more efficiently. While NDP is a relatively small protein, CFTR is a very large protein (1480 amino acid residues) consisting from several domains, that fold independently during translation35. Lower engagements of CFTR mRNA with polysomes may reflect slower translation that is required for cotranslational folding of its distinct domains.
Polysome fractions can be also used for detection of proteins. Detection of proteins in the gradient fractions was conducted on the example of ribosomal proteins in HeLa cells (Figure 4). Proteins were concentrated by precipitation with 10% TCA from the fractions and Western blot was used to detect the small subunit ribosomal protein RPS6 and large subunit ribosomal protein RPL11 (Figure 4,top panel). Their distribution correlated well with the distinct peaks on the absorbance spectrum. These experiments clearly demonstrate that the polysome fractions can be used to analyze proteins in them.
Many different sucrose concentrations gradients (for instance, 7-47%36, 5-50%7, 7-50%6 , 10-50%37, 15-50%8, and others) for polysomes fractionation were used. Here, we compared two gradients 10-50% and 17-51% (Figure 5). Although, 17-51% produced acceptable results, the separation in the 10-50% gradient was overall better.
It is well documented that chelating agents, such as EDTA, disrupt ribosomes and polysomes8,9. As it is shown in Figure 6, EDTA treatment of the HeLa lysate before loading on the gradient leads to disappearance of the peaks, corresponding to monosomes and polysomes, and significant increase in the ribosome subunits peaks. This experiment served as a control and demonstrated that the observed peaks without EDTA treatment are actually ribosomal monosomes and polysomes.
Figure 7 shows results of polysome fractionation from mouse testes. The absorbance spectrum has similarities with those from Leishmania and HeLa cells: distinct peaks of ribosomal subunits, monosomes and polysomes. Their shape and distribution produce a signature appearance, that make it easy to identify them on different polysomal spectra. Total RNAs were purified from the fractions and RNAs from selected fractions were analyzed by electrophoresis in agarose gel (Figure 7, top panel). The electrophoresis shows typical distribution of 18S and 28S ribosomal RNAs. Their sharp bands indicate intactness of the samples. The gel may be used for individual mRNAs detection by a following Northern blot or it can be used to evaluate quality of the samples before further experiments on RNA or protein analysis – the diffused ribosomal RNAs bands indicate RNA degradation in the samples.
During our studies, we used RNase inhibitor and Heparin as RNase inhibitors in the lysates and sucrose gradients. While both of them provided satisfactory results, the use of RNase inhibitor was preferable for RNA analysis because it does not inhibit the cDNA and RT-qPCR reactions. Thus, it did not require additional RNA purification steps. However, if researchers decide to use heparin during polysome preparation, be aware that heparin inhibits down-stream applications such as RT-qPCR and additional RNA purification step is needed (see Protocol section 6).
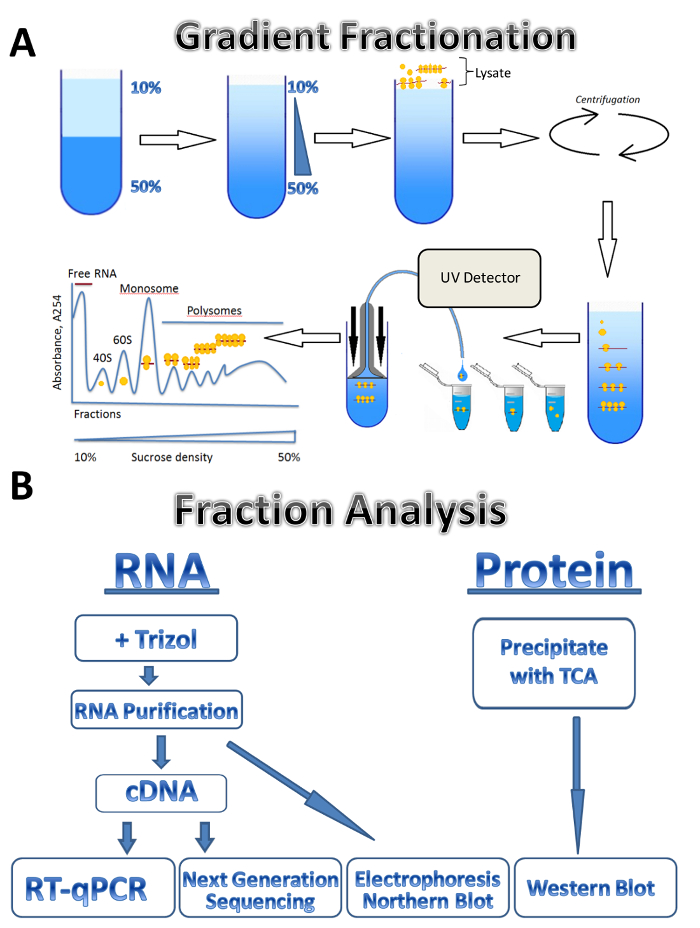
Figure 1. Polysome profiling. (A) Scheme of gradient preparation, polysome fractionation and absorbance profile. (B) Scheme of fraction analysis. Please click here to view a larger version of this figure.
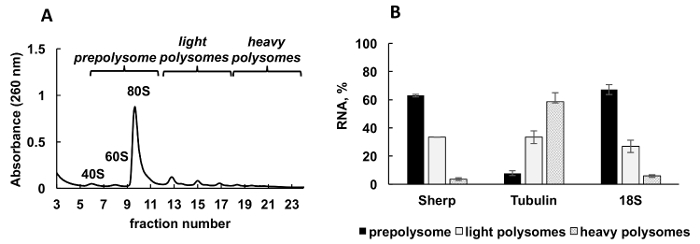
Figure 2. Polysome profile analysis of Leishmania major culture in logarithmic stage of growth. (A) Cytoplasmic lysate was fractionated in 10-50% sucrose gradient. (B) Relative distribution of 18S RNA, tubulin and Sherp mRNAs (%) in prepolysomes, light and heavy polysomes of log cells analyzed by RT-qPCR. Fractions containing 40S, 60S and monosomes were combined as prepolysomes. Fractions with 2-4 ribosomes were combined as light polysomes, while fractions with 5-8 ribosomes formed heavy polysomes. Synthetic E. coli OmpA mRNA added to fractions prior RNA extraction served as a normalization control in RT-qPCR. Comparative CT (ΔΔCT)31 method was used for calculation of mRNA levels. Error bars represent standard errors. Please click here to view a larger version of this figure.
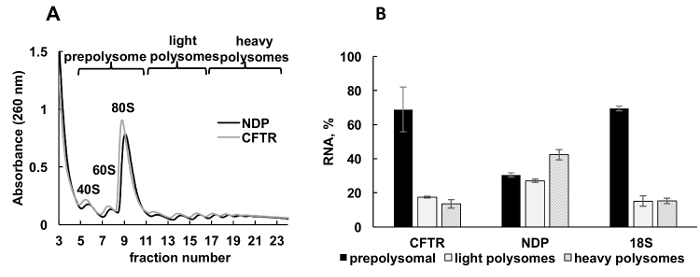
Figure 3. Polysome fractionation and analysis of recombinant CFTR and NDP mRNAs association with ribosomes in HeLa cells transfected with plasmid DNAs. (A) Polysomal profile in HeLa cells transfected with CFTR and NDP plasmids. 10%-50% sucrose gradient was applied to achieve separation of polysomes. The peaks for small (40S) and large (60S) subunits, as well as monosome (80S) are indicated. Fractions were combined as shown on panel A and used for further analysis. (B) Distribution of mRNAs of CFTR and NDP in different fractions. Detection of 18S by RT-qPCR was used as a control for polysome fractionation. RNA levels were evaluated by RT-qPCR analysis. Data were normalized using synthetic mRNA. Comparative CT (ΔΔCT)31 method was used for calculation of mRNA levels.Error bars represent standard errors. Please click here to view a larger version of this figure.
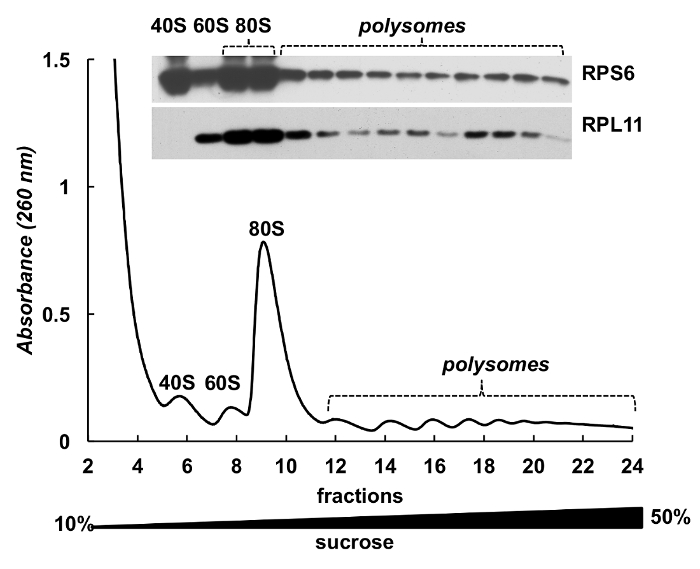
Figure 4. Detection of ribosomal proteins in HeLa polysomal fractions. HeLa cell lysate was subjected by 10%-50% sucrose gradient centrifugation. Proteins in selected fractions were precipitated with TCA and analyzed by electrophoresis in 12% SDS-PAGE with following Western blotting using mouse monoclonal RPS6 and rabbit polyclonal RPL11 antibody as primary antibodies and Peroxidase-Conjugated Goat anti-mouse or anti-rabbit secondary antibodies. Visualization of signals was done by SuperSignal West Pico PLUS chemiluminescent substrate. Please click here to view a larger version of this figure.
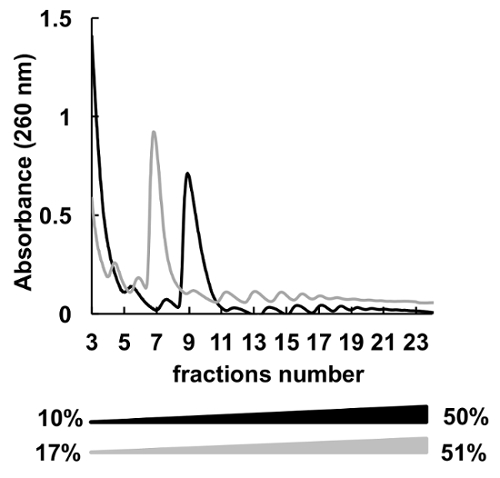
Figure 5. Comparison of HeLa polysomal profiling in 10%-50% (black) or 17%-51% (grey) sucrose gradients. Please click here to view a larger version of this figure.
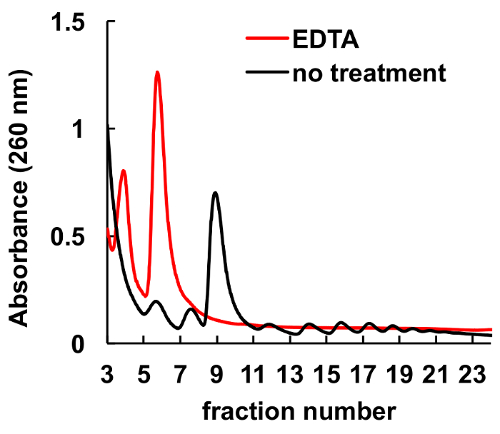
Figure 6. Effect of EDTA treatment on polysomal profile in HeLa cells. HeLa cell lysate was treated with 10 mM EDTA on ice for 10 min immediately before sucrose gradient centrifugation. MgCl2 was substituted by 5 mM EDTA in sucrose gradient solutions. Please click here to view a larger version of this figure.
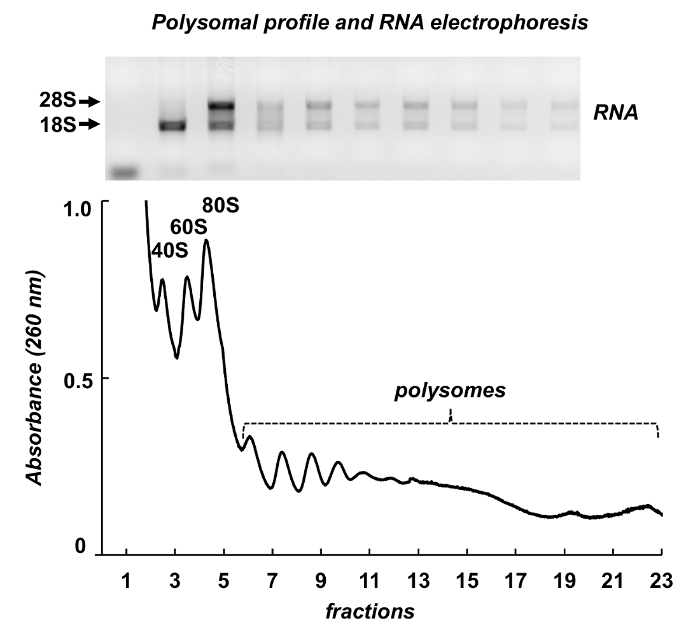
Figure 7. Polysomal profile from mouse testis tissue lysate. Fractions were subjected to RNA extraction with a RNA purification reagent and analyzed by electrophoresis in 1% agarose gel. Please click here to view a larger version of this figure.
| Stage | Step | Condition |
| Hold | Step 1 | Increase the temperature from 25 to 50°C with 1.6°C/s |
| Incubate at 50°C for 2:00 min | ||
| Step 2 | Increase the temperature from 50 to 95°C with 1.6°C/s | |
| Incubate at 95°C for 10:00 min | ||
| PCR | Step 1 | Incubate at 95°C for 00:15 min |
| Step 2 | Decrease the temperature from 95 to 60°C with 1.6°C/ | |
| Incubate at 60°C for 1:00 min | ||
| Number of cycles 40 | ||
| Melt Curve | Step1 | Increase the temperature from 60 to 95°C with 1.6°C/s |
| Step 2 | Decrease the temperature from 95 to 60°C with 1.6°C/s | |
| Incubate at 60°C for 1:00 min | ||
| Step 3 (dissociation) | Increase the temperature from 60 to 95°C with 0.05°C/s | |
| Incubate at 95°C for 00:15 min | ||
Table 1. Conditions for RT-qPCR
