General scheme
A general scheme of the protocol is shown in Figure 1. The protocol is divided into three major parts: fly work, sample preparation, sequencing and data analysis. The "Planning before the experiment" session of the protocol is not included in the general scheme for simplicity.
Timeline
A calendar of the major parts of the protocol (Fly work and Sample preparation) is shown in Figure 2.
Verifying cell-specific labeling by confocal microscopy
In a representative experiment, L3 lamina monopolar neurons (L3) were fluorescently labeled. L3 neuron is one of the five homologous lamina neurons (L1-L5) that receive input from the broadly tuned photoreceptors R1-R6 and relay the information to the high center by synapsing to target neurons in the medulla. In a MARCM experiment, single cell L3 clones are generated by mitotic recombination and fluorescently labeled by two markers, myr-tdTOM (membrane) and H2A-GFP (nuclear). The genotypes of flies used in the crosses are shown in Table 1. To verify cell-specific labelling, fly brains of the desired genotype were dissected at the developmental stage of interest (40 h APF). Immunostaining was performed as previously described7 using antibodies specific against dsRed and GFP (Figure 3, magenta and green), as well as the 24B10 antibody as a reference for the lamina and medulla neuropils (Figure 3, blue). Confocal microscopy confirmed that L3 is the only cell type labelled by both dsRed and GFP in the optic lobe.
Isolation of low-abundance cells by FACS
A representative FACS sorting result is shown in Figure 4. 100,000 events were recorded. 29.9% of all the events are potential singlets, and the rest could be debris or aggregates. Doublets and cells with different sizes were then gated out based on granularity and size. From the remaining single cells (FSC singlets, 27.3% of all events), L3 neurons appeared as a tight cluster in P1 (Figure 4, Specimen, magenta), which was well separated from the background cells (Figure 4, Specimen, blue). The size of the cells in P1 was similar consistent with a homogenous cellular population. Notably, a few cells in P2 (Figure 4, Specimen, green) with intermediate fluorescence intensity of dsRed and GFP were distributed between the tight cluster of cells in P1 and the background. The identity of these cells was not clear. Since P2 cells had a different size than the P1 cells, these cells were likely to be non-specific. To avoid potential contamination from the non-specific cells, only P1 cells were collected for the experiment. From 100,000 events, 45 P1 cells are obtained.
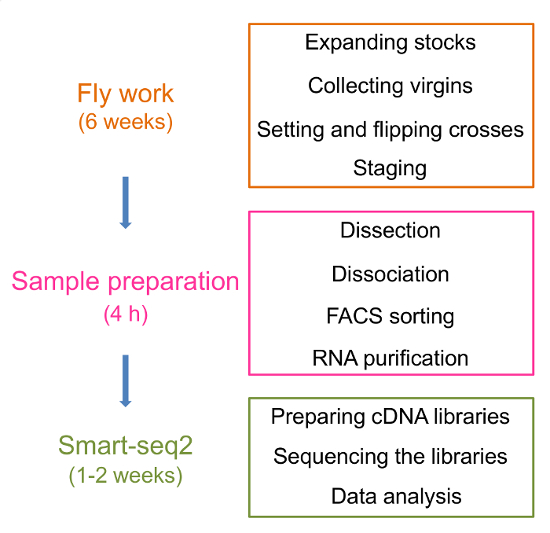
Figure 1: A general scheme of the protocol. The protocol consists of three parts: fly work, sample preparation, sequencing and data analysis. The approximate processing time of each part is indicated. Major steps of each part are also shown in the corresponding boxed regions. Please click here to view a larger version of this figure.
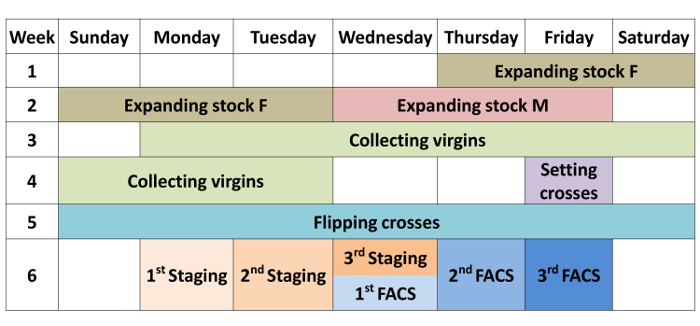
Figure 2: Timeline for fly work and Sample preparation. The protocol takes about 6 weeks. Timing for the major steps is shown in the calendar. Dissection, dissociation and RNA purification are done in the same day as the FACS sorting, and are not shown in the calendar for simplicity. Three biological replicates are done sequentially in the same week with the same crosses. Please click here to view a larger version of this figure.
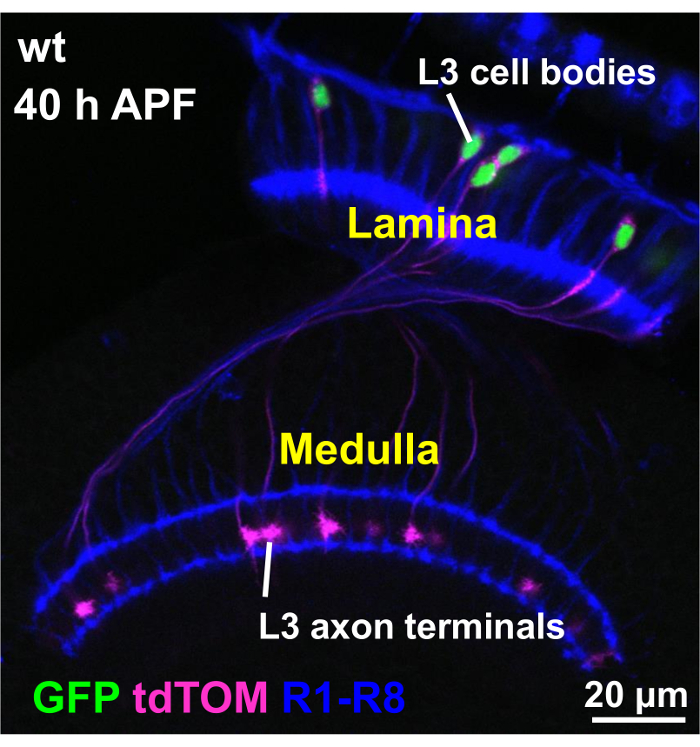
Figure 3: A representative confocal microscopy image showing the selective labeling of desired cells by fluorescent markers. In MARCM experiments, single L3 lamina monopolar neurons were made to express myr-tdTOM and H2A-GFP using an L3-specific GAL4 driver (9-9-GAL4)18. Fly brains of the desired genotype were dissected at 40hr APF and stained with anti-dsRed, anti-GFP and 24B10 antibodies (as a reference for the lamina and medulla neuropils). Fluorescent labeling was assessed by confocal microscopy. L3 neurons are born in the lamina and project axons that terminate within the medulla neuropil. In each brain, a subset of L3 neurons expressed both fluorescent reporters, and these were the only cells in the optic lobe expressing the markers. Please click here to view a larger version of this figure.
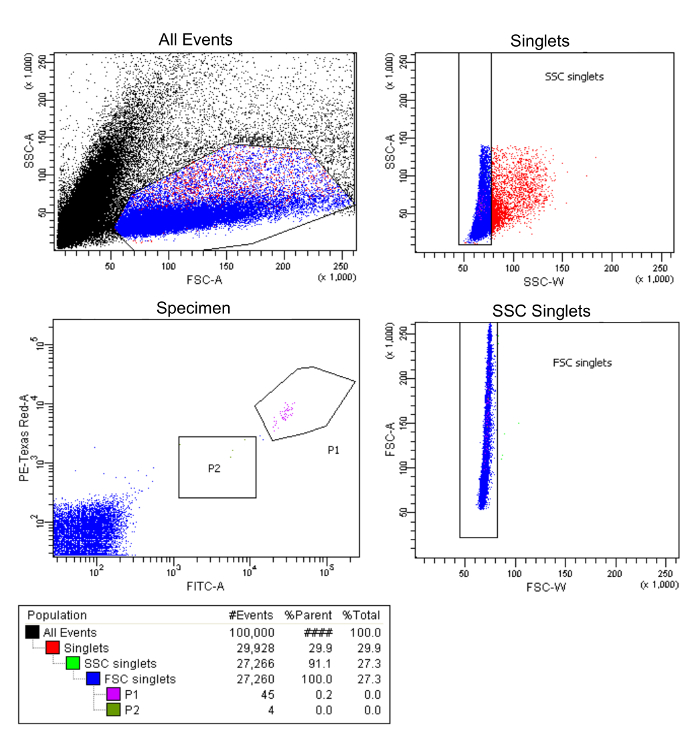
Figure 4: Purifying single L3 lamina neuron MARCM clones via FACS: a representative FACS plot. Gates (e.g. P1) were created based on cell granularity, size, and fluorescence intensity to isolate homogeneous single cells. L3 neurons expressing similarly high levels of myr-tdTOM and H2A-GFP were collected from in P1 (magenta). A few cells with intermediate fluorescence intensity are observed in P2 (green). These are different in size than L3 neurons in P1, and could represent non-specific cells. These were not collected. Please click here to view a larger version of this figure.
| Genotype | Source |
| w; TubP-GAL80, FRT40, 27G05-FLP::PEST/CyO, Kr-GAL4, UAS-GFP; 9-9-GAL4, UAS-myr::tdTOM/TM6B | Peng et al., 2018 |
| w; FRT40/CyO, Kr-GAL4, UAS-GFP; UAS-H2A-GFP/TM6B | Peng et al., 2018 |
Table 1: Genotypes of flies used in the crosses.
