DNA methylation is an important epigenetic mark that regulates gene expression and chromatin structure. Methylation occurs predominately in CpG dinucleotides — cytosine followed by guanosine; the methyl group is added to the 5-position of cytosine. Correct DNA methylation patterns, and thus proper gene expression, are needed for appropriate cellular development and function. Many disease states have been associated with changes to the normal methylation pattern1,2,3. For example, there is a link between cancer initiation and progression and alterations to the DNA methylation pattern. Typically, cancer cells exhibit lower overall levels of methylcytosine, which contributes to genome instability. At the same time, the methylcytosine that is present in the genome is concentrated in the promoter regions of tumor suppressor genes, which leads to gene silencing of these important proteins. Notably, epigenetic changes are dynamic and reversible, unlike the DNA mutations associated with tumorigenesis. This has made the proteins involved in epigenetic gene regulation interesting drug targets2,4.
DNA methyltransferases (DNMTs) are the proteins responsible for generating and maintaining DNA methylation patterns. Three catalytically active isozymes, DNMT1, DNMT3a, and DNMT3b, exist in humans. During development and differentiation, the de novo methyltransferases, DNMT3a and DNMT3b, establish methylation patterns. Both enzymes can bind the catalytically inactive DNMT3L protein to form complexes that exhibit increased activity1,5. Following cell division, daughter cells contain hemimethylated DNA — DNA containing methylcytosine in only one strand of the duplex — because the newly synthesized DNA is devoid of methylation marks. The major function of DNMT1 is to methylate this hemimethylated DNA, thus re-establishing the full methylation pattern1,5.
Links between DNMT activity and cancer are well established. Overexpression of DNMT1, either by transcriptional or post-translational mechanisms, is a consequence of several common oncogenic pathways6,7,8,9. Genetic approaches to lower DNMT1 activity using hypomorphic alleles result in decreased tumor formation in Apc(Min)mice10. Antisense oligonucleotides that knockdown DNMT1 inhibit neoplasia in cell culture and mouse tumor models11,12. Thus, inhibiting DNMT1 activity seems like a promising cancer therapy approach. However, the roles the DNMT3 isozymes play are not so straightforward. DNMT3a mutations are found in acute myeloid leukemia13 and myelodysplastic syndrome14. At least one of the identified mutations has been shown to decrease the DNA methylation activity of the enzyme15. However, DNMT3b is overexpressed in breast cancer16 and colorectal cancer17. With the various DNMT isozymes playing different roles in carcinogenesis, identifying isozyme-specific inhibitors will be critical. Not only will these compounds be useful for the development of therapeutics, but isozyme-specific inhibitors would also be an invaluable tool to dissect the role of each DNMT isozyme in cancer etiology.
Several DNMT inhibitors have been reported in the literature. Known DNMT inhibitors can be divided into two classes: nucleoside and non-nucleoside. Nucleoside inhibitors are typically cytidine analogs. These compounds are incorporated into DNA and covalently trap DNMTs. 5-azacytidine and 5-aza-2'-deoxycytidine have been approved for the treatment of myelodysplastic syndrome and acute myeloid leukemia4,18. The high toxicity, low bioavailability, and chemical instability of these compounds present problems. Ongoing work is examining the efficacy of the next generation of nucleoside inhibitors; SGI-110, derived from 5-aza-2'-deoxycytidine, is one example19,20. Nucleoside inhibitors are not isozyme-specific and will inactivate any DNMT isozyme encountered. Therefore, treatment with a nucleoside-demethylating agent results in the depletion of all DNMT isozymes4,18. Non-nucleoside inhibitors do not need to be incorporated into DNA to exert their inhibitory effects. Instead, these molecules bind directly to DNMTs, introducing the possibility for isozyme-specific inhibition. Several non-nucleoside inhibitors have been discovered to date, including SGI-102721, hydralazine22, procainamide23, RG108 and derivatives24, and natural products, (−)-epigallocatechin 3-gallate (EGCG)25 and laccaic acid A26,27. Most of the non-nucleoside inhibitors discovered to date are not isozyme-selective or display weak preferences for one DNMT isozyme. In addition, the potency of these molecules needs to be improved, especially in cells4,18. Thus, there is a need to discover or develop more potent, isozyme-selective DNMT inhibitors.
A hurdle to discovering new small molecule inhibitors of DNMTs is the laborious assays traditionally used to examine DNMT activity28. Assays are usually discontinuous with multiple steps. The enzymatic activity of DNMTs is still routinely assayed using radioactive S-adenosyl methionine (SAM)29,30,31,32,33,34. Non-radioactive assays for DNA methylation have been developed as well. For example, assays utilizing methyl-sensitive restriction endonucleases and electrophoresis to separate the digestion products have been described35,36. These types of discontinuous, multistep assays are not readily amenable to drug discovery. Since the mid-2000s, several DNA methylation assays with a higher throughput have been developed28. A scintillation proximity assay was used to screen for DNMT1 inhibitors37. Another assay utilizing a methyl-sensitive restriction endonuclease was used to screen for DNMT3a inhibitors25,38. While both assays allowed for higher throughput than traditional DNA methylation assays, the assays require multiple steps and do not allow the observation of methylation activity in real time. More recently, a continuous kinetics assay has been described that couples the formation of S-adenosylhomocysteine (SAH), one product of the methylation reaction, to the spectroscopic change at 340 nm associated with NADPH oxidation39. This assay utilizes three coupling enzymes to generate a spectroscopic signal.
We developed a fluorescence-based endonuclease-coupled DNA methylation assay that utilizes a single commercially available coupling enzyme and can generate data in real time (Figure 1). A hairpin oligonucleotide containing three methylcytosines is used as a substrate. The substrate DNA contains a fluorophore on the 5' end and a quencher on the 3' end. Methylation of the hemimethylated CpG site generates the cleavage site for the endonuclease Gla I — fully methylated GCGC. Gla I cleavage of the product oligonucleotide releases the fluorophore from the quencher and generates fluorescence in real time. The assay can be used to examine the activity of any isoform of DNMT; however, higher activity is observed with DNMT1 as this isozyme preferentially methylates hemimethylated DNA1,5. Even more robust activity is observed if the autoinhibitory Replication Foci Targeting Sequence (RFTS) domain is removed from DNMT1. This domain, found in the N-terminal regulatory region, binds to the catalytic site and prevents DNA binding. Removal of the first ~600 amino acids results in a truncated enzyme that is significantly more active than the full-length enzyme (~640-fold increase in kcat/Km)40. This activated form of the enzyme, referred to as RFTS-lacking DNMT1 (amino acids 621–1616), allows for the easier identification of inhibitors due to its increased catalytic power. This paper presents a protocol to utilize RFTS-lacking DNMT1 in assays to screen for potential small molecule inhibitors. Using the endonuclease-coupled continuous assay, the initial velocity is determined in the presence and absence of a few small molecules. Each potential inhibitor is examined at two concentrations to look for concentration-dependent DNMT1 inhibition. The percent activity observed in the presence of the small molecules was calculated in each case.
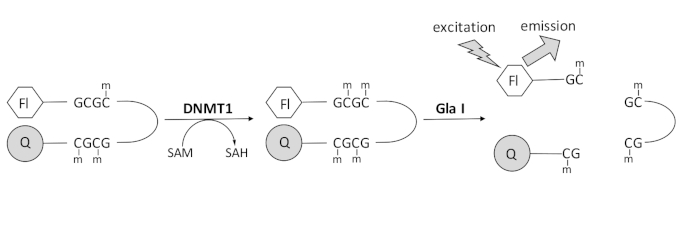
Figure 1: DNA methylation assay. A hemimethylated hairpin DNA with a fluorophore on the 5' end and a quencher on the 3' end is used as a substrate. DNMT1 catalyzes the transfer of the methyl group from S-adenosylmethionine to the nonmethylated CpG site, generating S-adenosylhomocysteine and fully methylated DNA. The DNA product contains the cleavage site for the endonuclease Gla I, which cleaves fully methylated GCGC sites. Cleavage of the product DNA releases the 5' fluorophore from the 3' quencher, generating fluorescence. Abbreviations: Fl = fluorophore; Q =quencher; DNMT1 = DNA methyltransferase 1; SAM = S-adenosylmethionine; SAH = S-adenosylhomocysteine. Please click here to view a larger version of this figure.
1. Prepare assay solutions for the screen
NOTE: The concentrations of substrates used in this assay can be adapted. For RFTS-lacking DNMT1, the experimentally determined Km values for the hairpin DNA substrate and SAM are 1–2 nM and 2 µM, respectively26,40.
- Prepare 600 µL of each assay condition being tested in microcentrifuge tubes on ice.
NOTE: The total volume of assay solution needed depends on the number of replicates being conducted. Each assay condition was run in triplicate for this protocol.- Add ddH2O and 5x methylation buffer (250 mM Tris pH 7.5, 5 mM MgCl2, 500 mM potassium glutamate, 5 mM dithiothreitol (DTT), 25% glycerol) to each tube to achieve a final concentration of 1x buffer. For assays containing 20 µM compound, add 472 µL of ddH2O and 120 µL 5x buffer. For assays containing 50 µM compound, add 470 µL of ddH2O and 120 µL of 5x buffer.
- Add 3.15 µL of 20 mg/mL bovine serum albumin (BSA) to each sample.
- Add 2 µL of 3.15 µM hairpin DNA substrate and 1.33 µL of 4.75 mM SAM to each sample.
NOTE: The concentration of substrates in the assay solution mixture is 1.05x the desired final concentrations. - Add inhibitor to a final concentration of 21 µM (1.26 µL of 10 mM) or 52.5 µM (3.15 µL of 10 mM) to the appropriate samples. Add an equivalent amount of dimethylsulfoxide (DMSO) to a control sample.
NOTE: The concentration of inhibitor used in the screen can be varied. The maximum final DMSO concentration should be ≤5% (v/v). DMSO concentrations up to 5% (v/v) have no effect on the activity observed in the assay26.
- Mix the solutions by vortexing (3,000 rpm) for 3 s. Spin the samples for a few seconds in a tabletop mini centrifuge (1,200 × g) to ensure that the solution is collected at the bottom of the tube.
- Aliquot 95 µL of each assay solution into 6 consecutive wells in a black half-area 96-well plate (Figure 2).
NOTE: These screening assays were performed in two batches. First, 20 µM inhibitor samples (4 total conditions) were examined, followed by the 50 µM inhibitor samples (4 total conditions).
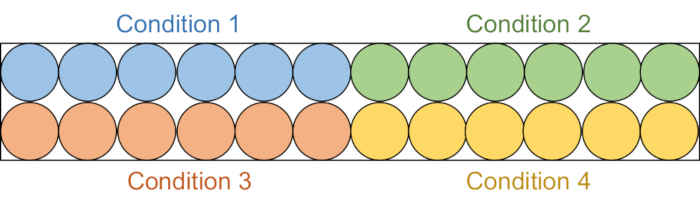
Figure 2: Assay plate setup. Each assay solution is aliquoted into six wells in the black 96-well plate: DMSO control (blue), compound 1 (green), compound 2 (red), and compound 3 (yellow). Both RFTS-lacking DNMT1 and Gla I will be added to three wells. As a control, Gla I alone will be added to the other three wells. Abbreviations: RFTS = Replication Foci Targeting Sequence; DNMT1 = DNA methyltransferase 1; DMSO = dimethylsulfoxide. Please click here to view a larger version of this figure.
2. Prepare enzyme solutions for screen
NOTE: RFTS-lacking DNMT1 can be expressed in E. coli and purified to homogeneity. Expression and purification procedures for RFTS-lacking DNMT1 have been described previously41. The volume of enzyme needed depends on the number of assays being conducted. Here, four different assays are being performed in each set; each assay is completed in triplicate.
- Prepare 75 µL of each enzyme solution in microcentrifuge tubes on ice.
NOTE: Two enzyme solutions will be needed. One solution will contain DNMT1 and Gla I; the other will contain only Gla I.- Add ddH2O and 5x methylation buffer (250 mM Tris pH 7.5, 5 mM MgCl2, 500 mM potassium glutamate, 5 mM DTT, 25% glycerol) to each tube to achieve a final concentration of 1x buffer. For the Gla I enzyme solution, add 15 µL of 5x buffer and 58.8 µL ddH2O. For the DNMT1+Gla I enzyme solution, add 15 µL of 5x buffer and 58.2 µL ddH2O.
- Add 1.2 µL of 10 U/µL Gla I to each solution for a final concentration of 0.16 U/µL.
- Add 0.6 µL of 5 µM RFTS-lacking DNMT1 for a final concentration of 40 nM to the DNMT1+Gla I solution.
- Tap gently to mix the solutions. Spin the samples for a few seconds in a tabletop mini centrifuge (1,200 × g) to ensure that the solution is collected at the bottom of the tube.
- Aliquot 12 µL of each enzyme solution into 6 wells in a conical-bottomed 96-well plate (Figure 3).

Figure 3: Enzyme plate setup. The Gla I (grey) and DNMT1+Gla I (blue) solutions are each aliquoted into six wells in the 96-well plate. Using a multichannel pipet, the enzyme can be added to a row of assay solutions simultaneously. For each assay condition (six wells), three wells will receive DNMT1+Gla I, and three wells will receive Gla I alone. Abbreviation: DNMT1 = DNA methyltransferase 1. Please click here to view a larger version of this figure.
3. Run the assay and analyze the data.
- Preheat the plate reader to 37 °C. Insert the black plate containing the assay solutions. Shake the plate (double orbital at 425 cpm for 5 s) and read the fluorescence with an excitation wavelength of 485 nm and an emission wavelength of 528 nm. Incubate the plate at 37 °C for 5 min.
NOTE: Alternatively, filters with proper wavelength cutoffs can be used for the fluorescence readings. - Remove the assay plate. Add 5 µL of enzyme solution to each well and mix by pipetting up and down.
NOTE: Use multichannel pipets so that a row of samples can be initiated simultaneously. - Insert the plate into the plate reader. Record fluorescence every 53 s for 30 min. Shake the plate (double orbital at 425 cpm for 4 s) before each reading.
- Average triplicate Gla I control assays for each condition. Subtract the average Gla I-containing control reaction trace from the triplicate traces obtained in the presence of RFTS-lacking DNMT1. Determine the average and standard error of the mean for the corrected replicates.
- Plot the average corrected reaction trace and fit the initial linear portion to a line to determine the initial velocity.
- Determine the percent activity by dividing the velocity observed in the presence of a compound by the velocity observed in the DMSO-containing control reaction and multiplying by 100.
4. Additional assay control — sequential addition of enzymes
- Prepare 550 µL of assay solution in a microcentrifuge tube on ice. Add 110 µL of 5x methylation buffer, 3.88 µL of 3.15 µM hairpin DNA substrate, 6.43 µL of 47.5 mM SAM, 3.06 µL of 20 mg/mL BSA, and 426.6 µL of ddH2O.
- Mix the solution by vortexing (3,000 rpm) for 3 s. Spin the samples for a few seconds in a tabletop mini centrifuge (1,200 × g) to ensure that the solution is collected at the bottom of the tube.
- Aliquot 90 µL of the assay solution into 6 consecutive wells in a black half-area 96-well plate.
- Prepare the DNMT1 enzyme solutions on ice. Add 4 µL of 5x methylation buffer, 0.76 µL of 5 µM RFTS-lacking DNMT1, and 15.24 µL of ddH2O to one tube. Add 4 µL of 5x methylation buffer and 16 µL of ddH2O to another tube.
- Tap gently to mix the solutions. Spin the samples for a few seconds in a tabletop mini centrifuge (1,200 × g) to ensure that the solution is collected at the bottom of the tube.
- Add 5 µL of the DNMT1-containing solution to three wells in the black half-area 96-well plate. Add 5 µL of the buffer control solution to the other three wells.
- Place the microtiter plate into the plate reader preheated to 37 °C. Shake the plate (double orbital at 425 cpm for 3 s) and read the fluorescence with an excitation wavelength of 485 nm and an emission wavelength of 528 nm. Repeat the shake and read every 60 s for 30 min.
- While the assay plate is in the plate reader, prepare the Gla I solution on ice. Add 8 µL of 5x methylation buffer, 0.64 µL of 10 U/µL Gla I, and 31.4 µL of ddH2O to a microcentrifuge tube. Tap gently to mix the solution. Spin the sample for a few seconds in a tabletop mini centrifuge (1,200 × g) to ensure that the solution is collected at the bottom of the tube.
- Aliquot 6 µL of the Gla I solution into 6 wells in a conical-bottomed 96-well plate.
- Following the initial 30 min read, remove the black plate from the plate reader. Add 5 µL of Gla I solution to all 6 wells and mix by pipetting up and down.
NOTE: Use a multichannel pipet so that all wells can be initiated simultaneously. - Place the black plate back into the plate reader. Record fluorescence every 35 s for 35 min. Shake the plate (double orbital at 425 cpm for 3 s) before each reading.
Active DNMT1 is a prerequisite for this analysis. RFTS-lacking DNMT1 was expressed in E.coli and purified to homogeneity following previously published procedures41. To ensure the purified enzyme was active, a discontinuous endonuclease-coupled assay was used to examine DNA methylation activity36. This assay utilizes a 32 base pair duplex DNA with a single hemimethylated CpG positioned in a Sau3A1 cleavage site. Sau3A1 can cleave the hemimethylated substrate DNA; however, cleavage is blocked when the DNA is fully methylated. RFTS-lacking DNMT1 was added to assays containing 1 µM DNA and 0.5 mM SAM, and aliquots were removed and flash-frozen over 30 min. Samples were subsequently digested with Sau3A1, and the products were separated by gel electrophoresis. As shown in Figure 4, the DNA is protected from cleavage as the reaction time increases, indicating that RFTS-lacking DNMT1 is methylating the hemimethylated DNA. Most of the 1 µM substrate DNA originally present in the assay has been converted to product over the 30-min time course.
The endonuclease-coupled assay described in this paper allows the observation of DNA methylation activity in real time as an increase in fluorescence. Key to the success of this assay is the methyl sensitivity of the endonuclease Gla I. Gla I does not efficiently cleave the hemimethylated substrate DNA but will cleave the fully methylated product DNA (Figure 1). Cleavage of the hairpin DNA is required to release the fluorophore from the quencher and generate an increase in fluorescence. To demonstrate that the enzymes are working as expected, a control reaction was conducted in which the enzymes, RFTS-lacking DNMT1 and Gla I, were added sequentially rather than concurrently. If the coupled assay is working correctly, the addition of DNMT1 should not impact fluorescence, as methylation of the DNA substrate is not expected to impact the background fluorescence of the internally quenched hairpin DNA. However, subsequent addition of Gla I to assays that contained the methyltransferase should result in fluorescence generation due to cleavage of the fully methylated hairpin DNA. The hemimethylated hairpin DNA substrate itself does have background fluorescence (Figure 5); these assays contained 20 nM hairpin DNA and 500 µM SAM. RFTS-lacking DNMT1 or buffer was added to the assays, and fluorescence was followed for 30 min.
As seen in Figure 5, the fluorescence is unaffected by RFTS-lacking DNMT1 in the absence of the coupling enzyme (blue shades contained 10 nM RFTS-lacking DNMT1 while red shades are the buffer control). Thus, methylation of the hemimethylated CpG site by DNMT1 does not appreciably change the fluorescence signal. However, when Gla I was added to all wells, robust fluorescence generation was observed only in the assays that contained RFTS-lacking DNMT1 (Figure 5). This shows that methylation of the hemimethylated substrate DNA by DNMT1 is required for Gla I cleavage and the generation of fluorescence. It should be noted that the increase in fluorescence observed in this control assay reflects the activity of Gla I as the enzymes were added sequentially. Alternatively, these reaction mixtures could be digested with Gla I, and the resulting oligonucleotides could be separated by electrophoresis to visualize cleavage and ensure that the enzymes are working as expected.
To use this coupled assay to directly determine the initial velocities of DNMT1, both enzymes, RFTS-lacking DNMT1 and Gla I, need to be added simultaneously. As long as the rate of Gla I cleavage of the product DNA is faster than the rate of DNA methylation by DNMT1, the fluorescence signal generated will reflect DNA methylation activity. To ensure that DNMT1 activity is rate-limiting, the concentration of DNMT1 can be varied while holding all other assay conditions constant. The rate of fluorescence generation should be proportional to the concentration of DNMT1. This has previously been shown for RFTS-lacking DNMT1 under the conditions utilized here40. When utilizing this coupled assay to examine DNMT activity, a Gla I-containing control reaction is performed in addition to the reaction that contains both DNMT1 and Gla I (Figure 6A). The Gla I control has several functions. Subtracting the Gla I control reaction trace from the DNMT-containing reaction trace allows for the accounting of both slow cleavage of the hemimethylated DNA substrate and the background fluorescence. The corrected reaction traces generated reflect the DNA methylation activity of DNMT1. Fitting the initial linear portion of the corrected reaction trace allows for the determination of the initial velocity (Figure 6B). With 10 nM hairpin DNA substrate and 10 µM SAM, an initial velocity of 113 ± 2 RFU/min was obtained.
Substituted anthraquinones have previously been shown to inhibit the activity of DNMT1. The natural product, laccaic acid A, a highly substituted anthraquinone, is a potent, DNA-competitive inhibitor of DNMT127. A few compounds with simpler structures, one or two substituents on the anthraquinone core, with one being a bulky aromatic substituent, have also been shown to inhibit DNMT1 in a DNA-competitive manner41. Here, the ability of three substituted anthraquinones or anthraquinone-like molecules to inhibit RFTS-lacking DNMT1 activity in the Gla I-coupled assay was examined as an example of how to conduct these screening assays. These compounds contained one to three substituents, all on one side of the anthraquinone core (Table 1). The substrate concentrations (10 nM DNA and 10 µM SAM) used for these assays are ~5x higher than the Km for each substrate. The signal generated in the assay comes directly from the substrate DNA. Because of this, it is difficult to assay at concentrations near or below Km; there is simply not enough signal to detect. These substrate concentrations were chosen because robust activity is seen under these conditions in the absence of inhibitors (Figure 6B). Other substrate concentrations could be used in screening assays. For example, conducting a screen at saturating SAM concentrations could be used as a strategy to bias the search against SAM-competitive inhibitors.
Each assay contained the hairpin DNA substrate, the methyl-donating co-factor SAM, and a potential inhibitor or DMSO as a control for the screen. We routinely generate 10 mM solutions of screening compounds in DMSO for use in assays. Anthraquinones such as those examined here exhibit poor solubility in aqueous solutions. Thus, to generate concentrated stocks, DMSO is used as a solvent. The addition of DMSO up to 5% (v/v) final concentration does not impact activity in the assay26. However, when dealing with compounds with low solubility in aqueous solution, care should be taken to ensure that the compound is soluble at the chosen final screening concentration(s).
For each condition examined in the screen, a Gla I-containing control assay is performed. In addition to accounting for background fluorescence and slow cleavage of the substrate hairpin DNA, this control allows the determination of whether the potential inhibitor interferes with the spectroscopic signal (e.g., it is fluorescent). After initiating the reaction by adding enzymes, fluorescence was measured for 30 min with a time interval of 53 s in the screening data. This is the fastest time interval available on the plate reader in this laboratory when reading an entire 96-well plate. Other time intervals could be used to generate the reaction traces. An appropriate time interval will depend on the instrument being utilized and the number of wells being read. To ensure reproducibility, each assay is conducted in triplicate. The average Gla I-containing control trace is subtracted from the reaction traces observed in the presence of RFTS-lacking DNMT1. The initial velocity of the reaction can be determined by fitting the initial linear portion of the corrected reaction traces.
Initially, the impact of three potential inhibitors on fluorescence generation was examined at a final concentration of 20 µM. Higher concentrations of the potential inhibitors were chosen for screening for two reasons. First, the substrate concentrations in the assay are both above their respective Km values. This can make it more difficult to detect inhibition in kinetics assays, particularly if the inhibitors are competitive. Second, the anthraquinone-like compounds used in this screen are significantly simpler in structure than the known inhibitor, laccaic acid A. As these molecules contain only a few substituents around the core structure, we anticipated weaker interactions. In the DMSO-containing control assay, an initial velocity of 111 ± 5 RFU/min was obtained (Figure 7A). Note, this is very similar to the rate reported earlier in the absence of DMSO and confirms that the addition of low amounts of DMSO does not impact the observed activity. The addition of two compounds decreased the observed initial velocity, while the addition of the third compound had no effect on activity. The initial velocities were used to determine the percent activity observed in the presence of each compound. At 20 µM, the addition of compounds 1 and 2 resulted in a percent activity of ~80%, while 100% percent activity was observed in the presence of compound 3, indicating that this molecule does not inhibit the enzyme (Table 1).
Inhibitors are expected to exhibit concentration-dependent inhibition. Next, these molecules were re-examined at an even higher concentration of 50 µM. For this set of assays, the DMSO-containing control assay had an initial velocity of 125 ± 7 RFU/min (Figure 7B). This velocity is slightly higher than the previously determined rates; however, these velocities are all relatively close within error. Again, compound 3 had little effect on the initial velocity, while the addition of compounds 1 and 2 decreased the observed initial velocity. As expected, at the higher concentration, compound 1 had a larger impact on activity. At 50 µM, the addition of compound 1 resulted in a percent activity of ~60% (Table 1). However, the addition of a higher concentration of compound 2 did not result in more robust inhibition. The percent activity observed in the presence of 50 µM of compound 2 was again near 80%.
As can be seen in the data presented, the endonuclease-coupled fluorescence-based DNA methylation assay can be used to screen for potential DNMT inhibitors. The data indicate that compound 1 and compound 2 are potential inhibitors, while compound 3 is not. Additional studies are required to validate the potential inhibitors as true DNMT1 inhibitors.
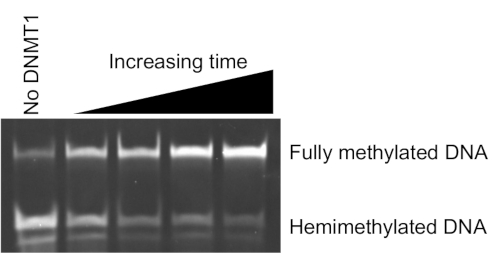
Figure 4: DNMT1 activity control. Hemimethylated duplex DNA methylation protects against Sau3A1 cleavage. RFTS-lacking DNMT1 was added to assay mixtures containing 1 μM DNA and 500 μM SAM. Samples were collected at the indicated time points during a 30-min incubation at 37 °C, digested with Sau3A1, and resolved by gel electrophoresis on 18% PAGE. Here, 5, 10, 15, and 30 min samples are shown. The first lane is a control lacking DNMT1. Abbreviations: DNMT1 = DNA methyltransferase 1; RFTS = Replication Foci Targeting Sequence. Please click here to view a larger version of this figure.
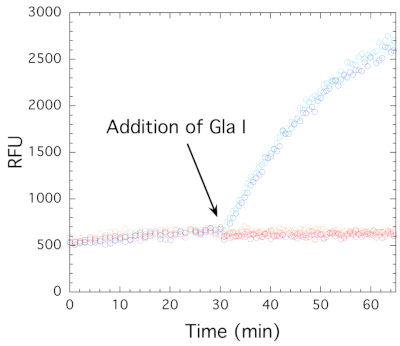
Figure 5: Fluorescence-based assay control. RFTS-lacking DNMT1 (10 nM final concentration) and Gla I (0.8 U) were added sequentially to triplicate assays containing 20 nM hairpin DNA substrate and 500 µM SAM. Addition of DNMT1 (blue circles) or buffer (red circles) alone had no effect on background fluorescence. Addition of Gla I to all assays results in fluorescence generation in assays that contained DNMT1, but not in assays that do not. Abbreviations: DNMT1 = DNA methyltransferase 1; RFTS = Replication Foci Targeting Sequence; RFU = relative fluorescence units. Please click here to view a larger version of this figure.
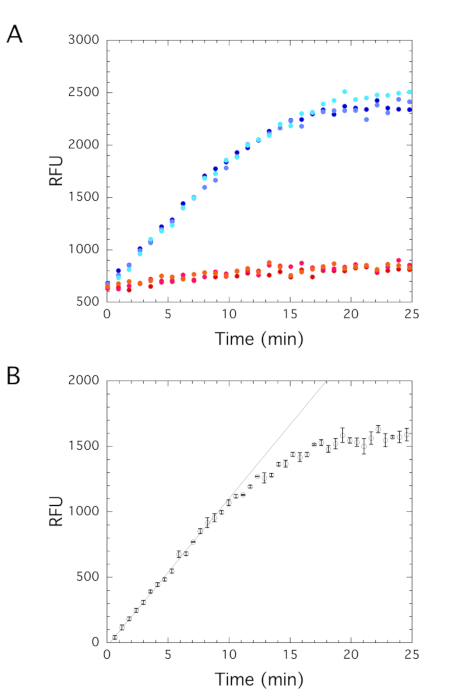
Figure 6: Fluorescence reaction traces. Triplicate assays contained 10 nM hairpin DNA substrate and 10 µM SAM. (A) RFTS-lacking DNMT1 (2 nM) and Gla I (0.8 U) were added to three assays (blue), and Gla I (0.8 U) alone was added to three assays (red). Robust fluorescence generation is observed only in the assays that contain both enzymes. (B) The average Gla I reaction trace was subtracted from the DNMT1+Gla I reaction traces. The average and standard error of the mean of the triplicate data are shown. Fitting the linear portion of the trace gives an initial velocity of 113 ± 2 RFU/min. Abbreviations: DNMT1 = DNA methyltransferase 1; RFTS = Replication Foci Targeting Sequence; RFU = relative fluorescence units; SAM = S-adenosylmethionine. Please click here to view a larger version of this figure.
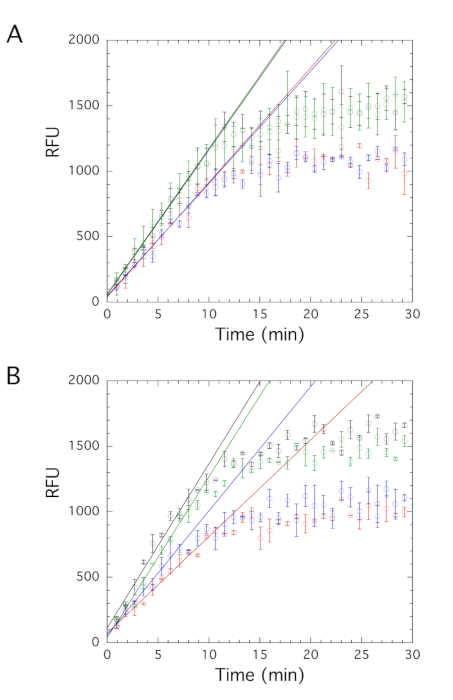
Figure 7: DNA methylation activity in the presence of potential inhibitors. DNA methylation activity of RFTS-lacking DNMT1 was examined in the presence of DMSO (black), compound 1 (red), compound 2 (blue), or compound 3 (green). Corrected reaction traces at 20 µM compound (A) and 50 µM compound (B) were fit to determine the initial velocity. Addition of compounds 1 and 2 decreased the observed velocity, while compound 3 had little impact on activity. Average and standard error of the mean for the triplicate assays are shown. Abbreviations: DNMT1 = DNA methyltransferase 1; RFTS = Replication Foci Targeting Sequence; RFU = relative fluorescence units; DMSO = dimethylsulfoxide. Please click here to view a larger version of this figure.
Table 1: Initial velocities and percent activities observed in the presence of potential small molecule inhibitors. Initial velocity was determined by fitting the initial linear portion of corrected reaction traces to a line; the error associated with the fit is reported. Percent activity was determined by dividing the rate determined in the presence of compound by the rate determined in the DMSO control reaction and multiplying by 100; associated error was propagated. Abbreviations: Cmpd = compound; DMSO = dimethylsulfoxide; RFU = relative fluorescence units. Please click here to download this Table.
