Het aantal kristalstructuren van eiwitten en eiwitcomplexen afgelopen jaren snel gestegen. Zij presenteren waardevolle momentopnamen van de structurele organisatie van deze eiwitten en vormen een basis voor de structuur-functie analyse. De dynamiek van eiwitten en conformationele veranderingen, die essentieel zijn voor hun functies, zelden onthuld door röntgenkristallografie. Cryo-elektronenmicroscopie, daarentegen, kan eiwitten en eiwitcomplexen vangen in verschillende conformaties, maar algemeen niet beneden oplossen conformationele veranderingen in secondaire structuur niveau 1. Conformationele dynamica van eiwitten in oplossing bij atomaire gegevens kunnen alleen worden opgelost door NMR, maar deze methode is gelimiteerd tot eiwitten relatief kleine grootte (gewoonlijk ≤ 30 kDa) en heeft hoge concentraties van eiwitten (≥ 100 uM), die experimenten belemmert met oligomerisatie of aggregatie gevoelig eiwitten 2. Een methode dieis in staat om een brug tussen hoge-resolutie X-ray kristallografie en cryo-elektronenmicroscopie en die wordt niet beperkt door proteïne grootte of concentratie amide waterstof-1 H / 2 H-uitwisseling (HX) in combinatie met massaspectrometrie (MS). De laatste jaren methode ontwikkeld om een waardevol analytisch instrument voor de analyse van eiwit dynamica, eiwitvouwing, eiwitstabiliteit en conformatieveranderingen 3-5. De moleculaire basis van deze methode is de labiele aard ruggengraat amide waterstof in eiwitten, die wisselen met deuterium atomen wanneer het eiwit in een D2O oplossing. De latere toename van eiwitmassa loop van de tijd wordt gemeten met een hoge-resolutie MS.
Kortom ongestructureerde peptiden HX alleen afhankelijk van de temperatuur, katalysatorconcentratie (OH -, H 3 O + dwz pH, zie figuur 3) en aminozuur-zijketens van aangrenzende residuen door inductieve, katkatalytische en sterische effecten. Deze effecten op de intrinsieke chemische wisselkoers k ch zijn elegant gekwantificeerd door Bai et al.. 6 en een programma beschikbaar is (met dank Z. Zhang), die k l berekent voor elk aminozuur in een polypeptide afhankelijk van pH en temperatuur. Bij neutrale pH en omgevingstemperaturen k l ligt in de orde van 10 1 -10 3 sec -1. In opgevouwen eiwitten HX kan 2-9 ordes van grootte trager vooral door waterstofbinding in de secundaire structuur en in mindere mate door beperkte toegang gehydrateerde OH – ionen om het interieur van een strak gevouwen eiwit. HX in natieve eiwitten impliceert dus gedeeltelijke of globale ontvouwen, chemische uitwisseling en opnieuw vouwen om de natuurlijke toestand volgens vergelijking (1) en de waargenomen wisselkoersen k obs afhangen van de opening tarief k op, de slotkoers k cl en de intrinsieke chemische uitwisseling rate k ch volgens vergelijking (2).
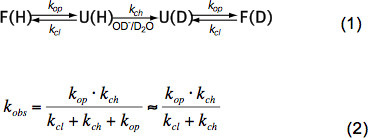
Onder natuurlijke toestand omstandigheden k op veel kleiner is dan k l en kan worden verwaarloosd in de noemer. Er zijn twee extreme uitwisseling regimes genoemd EX1 en EX2. Als de k cl is veel kleiner dan k l (EX1) is het waargenomen aantal is nagenoeg gelijk aan de opening tarief en HX staat directe observatie van het ontvouwen van een structureel element. Een dergelijke uitwisseling regime, waar alle amide protonen uitwisseling in een keer bij het openen van het structurele element, is gemakkelijk in MS door een bimodale verdeling van de isotoop pieken 7. Als k cl veel groter is dan k l (EX2) de waargenomen evenredig is met k ch waarbij de evenredigheidsconstante is gelijk aan het vouwen ontvouwen evenwichten k u = k op </sub> / K cl. Onder deze omstandigheden veel openen en sluiten gebeurtenissen nodig voordat alle amide protonen ruil voor deuteronen, leiden tot een geleidelijke stijging van de gemiddelde massa terwijl de isotopische verdeling blijft ongeveer gelijk. Het regime EX2 maakt de bepaling van de vrije energie van ontvouwing ΔG u en derhalve de stabiliteit van een constructiedeel. Onder natuurlijke toestand staat de EX2 regime is het meest gebruikelijk. Verhoging van de pH en de toevoeging van chaotrope middelen kan de uitwisseling mechanisme verschuiven naar EX1. Daarom kan HX-MS worden gebruikt thermodynamische staand en kinetische parameters van eiwitvouwing en conformatieveranderingen.
Zoals hierboven vermeld HX intrinsiek pH en temperatuur afhankelijke en uitwisseling halfwaardetijd van een geheel aan oplosmiddel blootgestelde proton van de hoofdketen amidegroep tussen 5-400 msec bij fysiologische pH (pH 7,6) en 30 ° C, maar 10 minuten naar> 15 uur met een gemiddelde van> 2 uur bij pH 2,9 en 0 °C (behalve het proton van de eerste ruggengraat amidebinding van een polypeptide, die wisselt met een halfwaardetijd van ca.. 1-2 min). Onder dergelijke trage uitwisseling omstandigheden is het mogelijk om het monster te verteren met proteasen (bijv. pepsine) die actief zijn onder deze omstandigheden, met uit verlies van alle in de opgenomen deuteronen gegevens. Sinds de introductie van peptische digestie onder langzaam uitwisselen omstandigheden kan niet alleen de totale HX kinetiek van full-length eiwitten geanalyseerd maar HX kan worden gelokaliseerd op specifieke gebieden 8,9. Ruimtelijke resolutie is momenteel beperkt tot de omvang van de peptische fragmenten, die in het algemeen tussen 10-30 residuen. Kan echter overlappende fragmenten te wijten aan de niet-specifieke aard van splitsing in pepsine tot een toename van ruimtelijke resolutie. Bovendien werden verscheidene andere proteasen actief bevonden onder quench omstandigheden echter veel minder efficiënt dan pepsine 10. Verdere increase van ruimtelijke resolutie kan worden bereikt door versnippering van peptiden in de gasfase door methoden die de deutereringsgraad patroon zoals electron capture dissociatie (ECD), elektron overdracht dissociatie (ETD) en infrarood multiphoton dissociatie (IRMPD) 11-13 bewaard. Deze technieken Teneinde het verlies van ruimtelijke resolutie door intramoleculaire proton migratie ("scrambling"), die wordt waargenomen door botsing geïnduceerde dissociatie (CID) de meest gebruikte techniek fragmentatie. Deze werkwijzen vereisen optimalisatie voor elke individuele peptide en is dus nog steeds zeer uitdagend.
HX-MS is gebruikt om eiwit-ligand en eiwit-eiwit interacties van virale capside assemblage 14-17 analyseren. Eiwit ontvouwen en opnieuw vouwen en de temperatuur geïnduceerde conformationele veranderingen werden onderzocht 7,18,19. Fosforylering en enkel aminozuur-mutatie-gerelateerde conformatie verandert 16,20 en nucleotide-geïnduceerde veranderingen werden geanalyseerd 21,22. Daarom lijkt deze methode bij uitstek geschikt voor montage en dynamica van moleculaire machines te analyseren. Een aantrekkelijke kandidaat, wiens mechanisme is van groot algemeen belang, is de Hsp90 chaperonne complex.
