El número de estructuras cristalinas de proteínas y complejos de proteínas aumentó rápidamente en los últimos años. Presentan instantáneas invaluables de la organización estructural de estas proteínas y proporcionan una base para el análisis de la estructura-función. Sin embargo, la dinámica de las proteínas y los cambios conformacionales, que son esenciales para sus funciones, rara vez se revelaron mediante cristalografía de rayos X. Cryo-microscopia electrónica, por otro lado, es capaz de capturar complejos de proteínas y de proteínas en diferentes conformaciones, pero en general no puede resolver cambios conformacionales a nivel de la estructura secundaria 1. Dinámica conformacional de proteínas en solución a detalles atómicos sólo pueden ser resueltas por RMN, pero este método todavía se limita a proteínas de tamaños relativamente pequeños (generalmente ≤ 30 kDa) y las necesidades de altas concentraciones de proteínas (≥ 100 micras), lo que dificulta experimentos con oligomerización o de agregación de proteínas propensas 2. Un método quees capaz de tender un puente entre la alta resolución de la cristalografía de rayos X y microscopía electrónica de crio-y que no está limitado por el tamaño de la proteína o la concentración es amida de hidrógeno-1 H / 2 H-intercambio (HX) en combinación con espectrometría de masas (MS). En los últimos años este método se ha desarrollado a una herramienta analítica valiosa para el análisis de la dinámica de proteínas, plegamiento de proteínas, estabilidad de la proteína y cambios conformacionales 3-5. La base molecular de este método es la naturaleza lábil de la cadena principal hidrógenos amida en proteínas, que se intercambio con átomos de deuterio cuando la proteína se coloca en un D2O solución. El posterior aumento de la masa de la proteína con el tiempo se mide con alta resolución de la EM.
En péptidos no estructurados cortos sólo HX depende de la temperatura, concentración de catalizador (OH -, H 3 O + es decir, pH, véase la Figura 3) y cadenas laterales de aminoácidos de residuos adyacentes debido a inductiva, gatoefectos alytic y estéricos. Estos efectos sobre el intrínseca tasa de intercambio químico K CH han sido elegantemente cuantificado por Bai et al. 6 y un programa está disponible (por cortesía de Z. Zhang), que calcula k CH para cada aminoácido dentro de un polipéptido dependiente de pH y la temperatura. A pH neutro y temperaturas ambiente K es CH en el orden de 10 1 -10 3 seg -1. En proteínas plegadas HX puede ser 2-9 ordenes de magnitud más lento debido principalmente a enlaces de hidrógeno en la estructura secundaria y en un grado menor debido al acceso restringido de hidratadas iones OH – al interior de una proteína fuertemente plegada. HX en las proteínas nativas, por tanto, implica desarrollo, intercambio químico parcial o global y replegado al estado nativo de acuerdo con la ecuación (1) y los tipos de cambio observados k obs dependen de la tasa de apertura op k, los k cl tasa de cierre y el intercambio químico intrínseca raTE CH K según la ecuación (2).
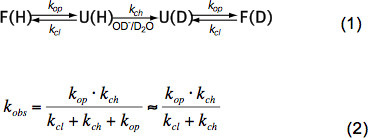
En condiciones de estado nativos K OP es mucho menor que K CH y se puede despreciar en el denominador. Hay dos regímenes cambiarios extremos llamados EX1 y EX2. Si el K CL es mucho menor que K CH (EX1) la tasa observada es prácticamente igual a la tasa de apertura y HX permite la observación inmediata del despliegue de un elemento estructural. Tal régimen de cambio, donde todos los protones amida de cambio a la vez después de la apertura del elemento estructural, es fácilmente observable en la EM por una distribución bimodal de los picos de los isótopos 7. Si k CL es mucho mayor que K CH (EX2) la tasa observada es proporcional a k CH mediante el cual la constante de proporcionalidad es igual a los equilibrios de plegado-desplegado constante K U = K OP </sub> K / cl. En estas condiciones, muchos eventos de apertura y cierre son necesarios antes de todo el intercambio de protones de amida de deuterones, dando lugar a un aumento gradual de la masa media, mientras que la distribución isotópica sigue siendo más o menos el mismo. El régimen EX2 permite la determinación de la energía libre de despliegue Delta G u y, por tanto, la estabilidad de un elemento estructural. Bajo condición de estado nativo régimen EX2 es más común. Aumento del pH y la adición de agentes caotrópicos puede cambiar el mecanismo de intercambio de EX1. Por lo tanto, HX-MS se puede utilizar para explorar termodinámico así como los parámetros cinéticos de plegamiento de la proteína y cambios conformacionales.
Como se mencionó anteriormente HX es intrínsecamente dependiente de pH y la temperatura y el cambio de la vida media de un protón completamente expuestos al disolvente del grupo amida del eje central es de entre 5-400 mseg a pH fisiológico (pH 7,6) y 30 ° C, pero 10 min a> 15 h con un promedio de> 2 horas a pH 2,9 y 0 °C (excepto para el protón de la primera enlace amida columna vertebral de un polipéptido, que intercambia con una vida media de ca. 1-2 min). Bajo tales condiciones lento intercambio es posible digerir la muestra usando proteasas (por ejemplo pepsina) que son activas en estas condiciones, con sin perder toda la información contenida en los deuterones incorporados. Desde la introducción de la digestión péptica en condiciones de intercambio lento, no sólo la cinética global HX de proteínas de longitud completa pueden ser analizados pero HX se pueden localizar en regiones específicas 8,9. La resolución espacial se limita actualmente al tamaño de los fragmentos generados pépticas, que es en general entre 10-30 residuos. Sin embargo, la superposición de fragmentos creados debido a la naturaleza no específica de la escisión por la pepsina podrían conducir a un aumento de la resolución espacial. Además, se encontraron varias otras proteasas para ser activa en condiciones de templado, sin embargo, mucho menos eficiente que la pepsina 10. Además increse de resolución espacial se puede llegar por la fragmentación de péptidos en fase gaseosa por métodos que conservan el patrón deuteración tales como la disociación de captura de electrones (ECD), la transferencia de electrones de disociación (ETD) y disociación multifotónica infrarroja (IRMPD) 11-13. Estas técnicas evitan la pérdida de la resolución espacial debido a la migración intramolecular de protones ("aleatorización"), que se observa por la disociación inducida por colisión (CID) la técnica de fragmentación más comúnmente utilizado. Sin embargo, estos métodos requieren la optimización para cada péptido individual y es por lo tanto todavía bastante difícil.
HX-MS se ha utilizado para analizar la proteína-ligando y proteína-proteína interacciones, incluyendo el montaje de la cápside viral 14-17. Proteína despliegue y replegamiento y se investigaron temperatura inducidas cambios conformacionales 7,18,19. La fosforilación y solo aminoácido conformacional relacionados mutación cambia 16,20 y nucleotSe analizaron los cambios ide-inducidos 21,22. Por lo tanto, este método parece idealmente adecuado para analizar el montaje y la dinámica de máquinas moleculares. Un candidato atractivo, cuyo mecanismo es de gran interés general, es el complejo chaperona Hsp90.
