The cell generates traction forces on the substrate by its focal adhesions. Force generated by cytoskeletal elements are in equilibrium with the counteracting force of the culture substrate. After laser induced lesion of the neurite, some of the tensed cytoskeleton cables are disrupted and their equilibrated tension is released because the opposing force of the substrate adhesion is eliminated. The released tension is partially distributed on the unaffected focal adhesions, and the bead attached to the cell membrane, held in an optical trap, measures the portion of such release not counteracted by cytoskeletal elements anchoring the cell to the substrate (see schematic Figure 1).
We report, in Figure 2, a representative result of the above-described experimental protocol. The neuron, in Figure 2a left panel, presents a longer neurite, which identifies the axon of the differentiating neuron. In Figure 2a right panel, the same neuron is shown after the induced axonal lesion (indicated by the white arrow). Figure 2b shows the recorded bead displacement traces in x, y, and z directions. Figure 2c reports the video tracking of a scratch on the glass support, to take into account and eliminate the stage drift during the measurement.
In Figure 3a, on the left panel, we show the lesioned neurite, and the white line upstream the lesion site, where we calculate the kymograph of the neurite diameter during the tension release measurement. On the right panel is the kymograph, showing how the neurite diameter slightly increases immediately after the lesion, and then becomes thinner due to the release of tension toward the cell soma. In Figure 3b, we quantify the kymograph result. The neurite appears brighter with respect to the background since we positioned the attached bead in the focus center of the objective, and therefore the axon is slightly out of focus. Indeed, by reporting the sum of pixel intensities along the kymograph line at each frame of the time-lapse video recording, we obtain an estimate of the neurite diameter during the release of tension. On panel 3c, we report the total variance of the attached bead during the tension release, calculated based on the traces recorded by the interferometer (in Figure 2b) after high pass filtering at 10 Hz cut-off frequency. We can observe that the variance increases with the neurite diameter during accumulation of material upstream of the lesion site. Then, the variance starts to decrease (at t0 in Figure 3c) when the membrane is strained, and the neurite diameter decreases too. The variance decrease reaches a plateau (at t1 in Figure 3c), when the tension release ceases. After t1, the Brownian motion starts to increase because of membrane relaxation possibly due to exocytosis21 (see Figure 3c).
In Figure 4a, the white box indicates the estimation of the neurite contact area Aaxon with the culture support. Aaxon is calculated between the UV laser spot position and the center of the trapped probe. Figure 4b reports the amplitude trace of the total released force Freleased obtained after multiplying the drift-corrected QPD displacement traces (in Figure 2b) by the respective calibrated optical trap stiffness (kx, ky, kz). We calculate the difference between force measured at time t1 and t0 (ΔpN in Figure 4b), and then we divide by Aaxon, to obtain the tension released in terms of pN/μm2.
By repeating the same protocol on different neurons in the sample, we can start sessions of experiments on several cultures treated with chemical factors or plated on different supports, and finally compare the average value obtained in the distinct experimental conditions. In Figure 5, we show the release of tension after axonal lesion is dampened by focal adhesions on the substrate, thus a higher tension release value means an axon less adhered to the substrate. Because we induce a partial, and not complete, lesion of the axon, we investigated the dependence of the measured tension release on the total energy delivered, and on the neurite contact area between the lesion site and the trapped bead. We sought to destroy more cytoskeletal elements by delivering a higher number of light pulses, and consequently induce a higher release of tension. Otherwise, with an increased neurite contact area, we expected to measure a lower amount of tension release. In Figure 5, we show that the dependency on the two above-mentioned parameters is not clear. Therefore, we deduced that the induced partial lesion (with the previously calibrated low energy per pulse; see protocol step 4.5), is related to the dimension of the ablation spot8, which does not vary when using the same microscope objective and the same energy per pulse at the sample. Moreover, the fact that the release of tension is not correlated to the contact area is not surprising because it is well known that such parameters do not represent a good estimate of the cell adhesion to the substrate, as focal adhesions occupy only 1% of the basal surface22.
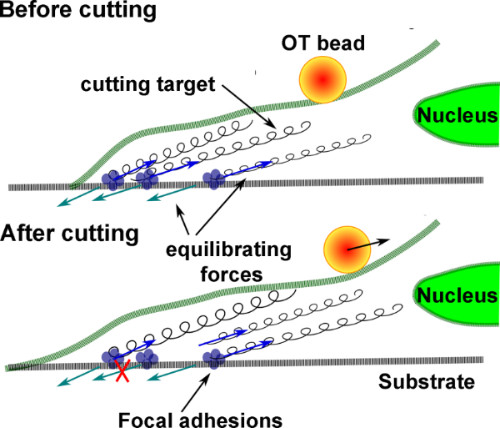
Figure 1. Graphical representation of the disrupted cytoskeleton equilibrium during laser axotomy. A cell adheres to a substrate by focal adhesions. Blue arrows specify traction forces generated by cytoskeletal elements (indicated by springs). Light-blue arrows indicate the counteracting forces generated by the rigid culture substrate. In a simplified scenario, before lesion, the cytoskeletal and substrate forces are paired and equilibrated. After laser-induced lesion of the neurite, the connections between some springs and the substrate are disrupted. Thus, the substrate is no longer counteracting the traction force of the cytoskletal element. The trapped bead, attached to membrane, tracks the direction of the released cytoskeleton forces.
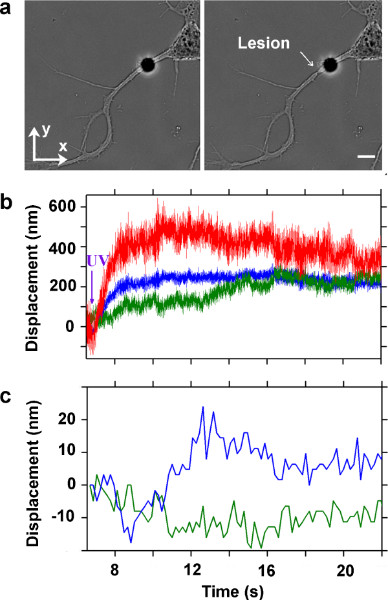
Figure 2. Interferometric tracking of a trapped bead, attached to the membrane of the axon of a hippocampal neuron during laser induced lesion. (a) Bright field images acquired during ablation of an axon. Before the lesion on the left panel. After the lesion on the right panel. A poly-D-Lysine coated bead is attached to the membrane (polystirene bead, Ø 4 μm) and held in an optical trap. Average power of the IR laser at the sample is 14 mW. White arrow indicates the lesion site. Bar is 5 μm. (b) Recorded traces by back focal plane interferometry of the bead position in the optical trap. Blue, green and red traces represent the bead position along x, y, and z axes, respectively. Sampling rate is 20 kHz. (c) Displacement of a scratch on the culture support measured by video tracking to monitor the stage drift. Blue and green traces are the x and y positions obtained by video tracking. Frame rate is 5.5 Hz.
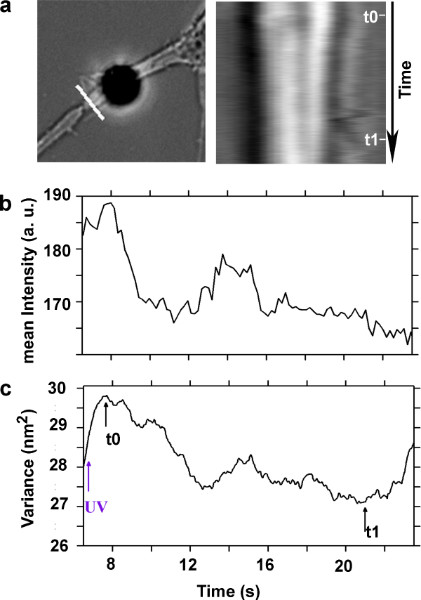
Figure 3. Analysis of the axon diameter during the tension release, and the membrane strain of the lesioned axon. (a) Bright field image of the lesioned axon, on the left panel. The white line perpendicular to the axon indicates the position where a kymograph is computed. On the right panel, kymograph of the axon diameter, upstream of the lesion site. Each row of the kymograph corresponds to 0.18 sec. (b) Sum of pixel intensities of each row of the kymograph representing an estimate of the axon diameter during the tension release. (c) Total variance of the Brownian motion of the bead attached to the axon (t0 and t1 indicate respectively the beginning and the end of tension release in the axon after dissection).
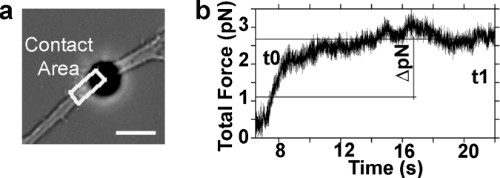
Figure 4. Quantification of the tension released after axotomy. (a) Bright field image of the lesioned axon. The white box indicates the estimate of the neurite contact area between the lesion site and the center of the trapped bead. (b) Trace of the total amplitude of the force measured after axotomy (Ftot= √(Fx2+Fy 2+Fz 2)). Fx, Fy, and Fz were calculated by Hooke’s law (F = kx), where the displacement traces are the ones shown in Figure 2b. The stiffnesses, in the three orthogonal directions, of the optical trap were calculated by the power spectrum method (kx,y = 7.9 pN/μm, kz = 2.3 pN/μm). ΔpN indicates the measured released force between the time instants t0 and t1.
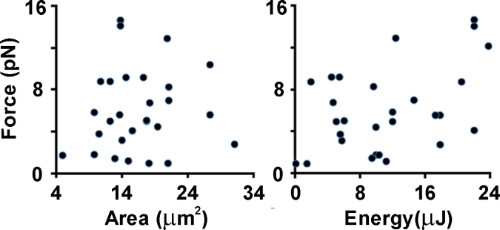
Figure 5. Dependence of measured tension release on the amount of energy delivered to the sample, and on the neurite contact area. Data from 26 experiments performed on the axons of mouse hippocamapal neurons at 3 DIV. (a) Scatter plot of tension release versus neurite contact area between lesion and trapped bead locations. (b) Scatter plot of tension release versus total amount of energy delivered to the sample neurite contact area.
