Le nombre de structures cristallines des protéines et complexes de protéines a augmenté rapidement ces dernières années. Ils présentent des instantanés précieux de l'organisation structurelle de ces protéines et fournissent une base pour l'analyse structure-fonction. Cependant, la dynamique des protéines et les changements de conformation, qui sont essentielles pour leurs fonctions, sont rarement révélées par cristallographie aux rayons X. Cryo-microscopie électronique, d'autre part, est capable de capturer des complexes de protéines et des protéines dans des conformations différentes, mais en général ne peut pas résoudre les changements conformationnels jusqu'au niveau de la structure secondaire 1. Dynamique conformationnelle des protéines en solution à des détails atomiques ne peuvent être résolus par RMN, mais cette méthode reste limitée aux protéines de taille relativement petite (généralement ≤ 30 kDa) et les besoins des concentrations élevées de protéines (≥ 100 de pM), qui entrave expériences avec oligomérisation ou d'agrégation des protéines sujettes 2. Une méthode quiest capable de combler entre haute résolution cristallographie aux rayons X et cryo-microscopie électronique, et qui n'est pas limitée par la taille de la protéine ou de la concentration de l'hydrogène est l'amide-1 H / 2 H-échange (HX) en combinaison avec la spectrométrie de masse (MS). Ces dernières années, ce procédé a mis au point un outil d'analyse utile pour l'analyse de la dynamique des protéines, le repliement des protéines, la stabilité des protéines et des changements de conformation 3-5. La base moléculaire de cette méthode est la nature labile du squelette hydrogènes d'amide dans les protéines, qui vont échanger avec des atomes de deutérium lorsque la protéine est placé dans une solution de D 2 O. L'augmentation subséquente de la masse de la protéine au cours du temps est mesurée avec MS à haute résolution.
En bref peptides non structurés ne HX dépend de la température, la concentration en catalyseur (OH -, H 3 O + ie pH, voir la figure 3) et des chaînes latérales d'acides aminés des résidus adjacents dues à inductive, chateffets alytic et stériques. Ces effets sur la valeur intrinsèque du taux de change chimique k ch ont été élégamment quantifiée par Bai et al. 6 et un programme est disponible (avec la permission Z. Zhang), qui calcule k ch pour chaque acide aminé dans un polypeptide dépend du pH et de la température. A pH neutre, et des températures ambiantes ch k est de l'ordre de 10 1 à 10 3 s -1. Dans les protéines pliées HX peut être 2-9 ordres de grandeur plus lentes principalement due à la liaison hydrogène dans la structure secondaire et à un degré moindre en raison de l'accès restreint de hydratés ions OH – à l'intérieur d'une protéine étroitement replié. HX en protéines natives implique donc déroulement, l'échange chimique partielle ou globale et repliement à l'état natif selon l'équation (1) et les taux de change observés k obs dépendent du taux d'ouverture k op, les taux de clôture k cl et l'échange chimique intrinsèque rate ch k selon l'équation (2).
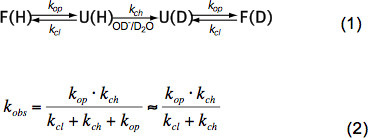
Dans des conditions natives de l'Etat k op est beaucoup plus petit que k ch et peut être négligée dans le dénominateur. Il existe deux régimes de change extrêmes appelés EX1 et EX2. Si la cl k est beaucoup plus petit que k ch (EX1) du taux observé est pratiquement égale à la vitesse d'ouverture et de HX permet l'observation immédiate du déploiement d'un élément de structure. Un tel régime de change, où tout protons amide échange à la fois lors de l'ouverture de l'élément structurel, est facilement observable dans MS par une distribution bimodale des pics isotopiques 7. Si k cl est beaucoup plus grand que k ch (EX2) du taux observé est proportionnel à k ch lequel la constante de proportionnalité est égal aux équilibres de pliage-dépliage constante K = k u op </sub> / K cl. Dans ces conditions, beaucoup d'ouverture et de clôture des événements sont nécessaires avant tout échange de protons amide pour deutons, conduisant à une augmentation graduelle de la masse moyenne, tandis que la distribution isotopique reste à peu près le même. Le régime de EX2 permet la détermination de l'énergie libre de dépliement Ag u et donc la stabilité d'un élément de structure. Sous la condition native de l'Etat du régime de EX2 est la plus courante. Augmentation du pH et l'ajout d'agents chaotropiques peut changer le mécanisme de change pour EX1. Par conséquent, HX-MS peut être utilisée pour explorer thermodynamique ainsi que des paramètres cinétiques de repliement des protéines et des changements conformationnels.
Comme mentionné ci-dessus HX est intrinsèquement dépendante du pH et de la température et de l'échange de demi-vie d'un proton complètement exposé au solvant du groupe amide du squelette est comprise entre 5 à 400 msec à pH physiologique (pH 7,6) et 30 ° C, mais à 10 min> 15 h avec une moyenne de> 2 h à pH 2,9 et à 0 °C (sauf pour le proton de la première épine dorsale liaison amide d'un polypeptide, qui échange avec une demi-vie de ca. 1-2 min). Dans ces conditions, l'échange lent, il est possible de digérer l'échantillon en utilisant des protéases (par exemple la pepsine) qui sont actives dans ces conditions, avec les perdre toutes les informations contenues dans les deutons incorporés. Depuis l'introduction de la digestion gastro-duodénal dans des conditions échanger lents, non seulement la cinétique de HX ensemble de protéines de pleine longueur peuvent être analysés mais HX peuvent être localisés à des régions spécifiques 8,9. La résolution spatiale est limitée à la taille des fragments peptiques générés, ce qui est en général comprise entre 10-30 résidus. Cependant, les fragments chevauchants créés en raison de la nature non spécifique de clivage par la pepsine pourraient conduire à une augmentation de la résolution spatiale. En outre, plusieurs autres proteases se sont révélés être actifs dans des conditions de trempe, cependant, beaucoup moins efficaces que la pepsine 10. En outre augsoi de la résolution spatiale peut être atteint par la fragmentation de peptides en phase de gaz par des méthodes qui ont préservé le modèle de deutération comme la capture d'électrons dissociation (DPE), transfert d'électrons dissociation (ETD) et infrarouge dissociation multiphotonique (IRMPD) 11-13. Ces techniques empêchent la perte de résolution spatiale due à la migration intramoléculaire de proton ("brouillage"), qui est observée par dissociation induite par collision (CID) de la technique de fragmentation le plus couramment utilisé. Cependant, ces méthodes nécessitent l'optimisation pour chaque peptide individuel et est donc encore assez difficile.
HX-MS a été utilisé pour analyser les interactions protéine-ligand et la protéine-protéine, y compris l'assemblage de la capside virale 14-17. Protéines de repliage et dépliage ainsi que la température des changements conformationnels induits ont été étudiés 7,18,19. Phosphorylation et unique de conformation liée à la mutation d'acides aminés et modifie 16,20 nucleotchangements ide-induits ont été analysés 21,22. Par conséquent, cette méthode semble parfaitement adapté pour analyser l'assemblage et la dynamique des machines moléculaires. Un candidat intéressant, dont le mécanisme est d'un grand intérêt général, est le complexe chaperon Hsp90.
