단백질 및 단백질 복합체의 결정 구조의 수는 최근에 급격히 증가 하였다. 그들은 이러한 단백질의 구조적 조직의 귀중한 스냅 선물 구조 – 기능 분석을위한 기초를 제공한다. 그러나, 단백질의 기능에 필수적인 구조적 변화의 역학은 거의 X-선 결정학에 의해 공개되지 않습니다. 극저온 electronmicroscopy, 반면에, 상이한 입체 형태로 단백질 및 단백질 복합체를 캡처 할 수 있지만, 일반적으로 이차 구조 레벨 1에 형태 적 변화를 해결할 수 없다. 원자 세부 사항 용액에 단백질의 구조적 역학은 NMR에 의해 해결 될 수 있지만,이 방법은 여전히 상대적으로 작은 크기 (일반적으로 ≤ 30 kDa의)의 단백질로 제한하고 실험을 방해하는 단백질의 높은 농도 (≥ 100 μM)를 필요로한다 올리고머 또는 집계 발생하기 쉬운 단백질 2. 한 가지 방법이고해상도의 X-선 결정학 및 크라이 electronmicroscopy 어느 사이 해소 할 수는 단백질의 크기 나 농도에 의해 제한되지 않는다 아미드 수소 한 질량 분석계 (MS)와 결합 H / 2 H 교환기 (HX)입니다. 최근 몇 년 동안이 방법은 단백질 역학, 단백질 접힘, 단백질의 안정성과 구조적 변화 3-5의 분석을위한 유용한 분석 도구를 개발했습니다. 이 방법의 분자 기초는 단백질 D 2 O 용액에 배치 될 때, 중수소 원자와 교환 할 것이다 단백질 백본 아미드 수소의 불안정한 성질이다. 시간에 따른 단백질 질량의 후속 증가는 고해상도 MS로 측정된다.
짧은 비정형 펩티드 만 온도, 촉매 농도에 따라 HX (OH -도 3 참조, H 3 O + 즉 산도) 및 유도, 고양이에 의한 인접 잔기의 아미노산 측쇄alytic 및 입체 효과. 고유 화학 환율 K의 채널에 이러한 효과는 우아 바이 등. (6)에 의해 정량화하고 프로그램은 pH와 온도에 따라 폴리펩티드 내의 각 아미노산 K의 채널을 계산 (예의 Z. 장), 사용할 수 있습니다. 중성 pH 및 주위 온도에서 K의 채널 10 1 -10 3 초 -1의 순서입니다. 단단히 접힌 단백질의 내부에 이온 – 접힌 단백질 HX는 주로 이차 구조의 수소 결합과 OH 수화의 접근 제한으로 인해 작은 정도 느리다 2-9 명령 할 수있다. 천연 단백질 HX 따라서 일부 또는 글로벌 전개, 화학 교환 및 식에 따라 기본 상태로 폴딩 연루 (1)과 K OBS 여는 속도 K의 영업 이익, 마감 환율 케이의 CL과 고유 화학 교환에 따라 관찰 환율 라TE K CH 식에 따라 (2).
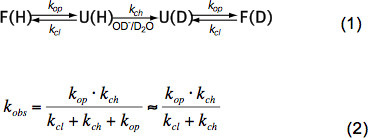
원시 상태의 조건에서 K의 영업 이익은 K의 채널보다 훨씬 작고 분모에서 무시 될 수있다. EX1과 EX2라는 두 개의 극단적 인 환율 제도가 있습니다. K 개의 CL은 K 개의 CH (EX1)보다 훨씬 작은 경우 관찰 속도는 개구율 실질적 같고 HX는 구조 부재의 전개를 즉시 관찰 할 수있다. 구조 요소의 열 때 한 번에 모든 아미드 양성자 교환, 동위 원소 피크 (7)의 바이 모달 분포에 의해 MS에서 쉽게 관찰 할 수 이러한 교환 정권. K의 CL은 K의 채널보다 훨씬 큰 경우 비례 상수는 접이식 전개 equilibriums에 해당된다 (EX2)는 관찰 된 비율은 K의 채널에 비례 상수 K U = K 영업 이익 </sub> / K CL. 동위 원소의 분포가 거의 동일하게 유지하면서 이러한 조건에서, 많은 개폐 이벤트는 평균 질량의 점진적인 증가로 이어지는, deuterons에 대한 모든 아미드 양성자 교환하기 전에 필요합니다. EX2 정권은 ΔG U 전개의 자유 에너지의 결정과 구조적 요소 때문에 안정성을 할 수 있습니다. 원시 상태의 조건에서 EX2 정권은 가장 일반적입니다. pH와 카오 트로픽 요원의 추가의 증가는 EX1에 교환 메커니즘을 전환 할 수 있습니다. 따라서, HX-MS는 열역학적을 탐구뿐만 아니라 단백질 접힘 및 구조적 변화의 운동 매개 변수를 사용할 수 있습니다.
HX 위에서 언급 한 바와 같이하는 것은 본질적으로 pH와 온도에 의존하고 백본 아미드 기의 완전히 용매 노출 된 양성자 교환 반감기>에 생리적 pH에서 5-400 밀리 초 (산도 7.6), 30 ° C,하지만 10 분 사이 pH가 2.9에서> 2 시간의 평균과 0 °와 15 시간(캘리포니아의 반감기. 1 ~ 2 분으로 교환하는 폴리 펩타이드의 첫 번째 백본 아미드 결합의 양자 제외) C. 느린 교환 조건 하에서 아웃이 혼입 deuterons에 포함 된 모든 정보를 잃고,이 조건 하에서 활성 단백질 분해 효소 (예 : 펩신)를 사용하여 샘플을 소화 할 수있다. 느린 교환 조건에서 소화 소화의 도입 이후, 전체 길이 단백질의 전체 HX 반응 속도뿐만 아니라 분석 할 수 있지만, HX는 특정 지역 8,9에 지역화 할 수 있습니다. 공간 해상도는 현재 10 ~ 30 잔기 사이에 일반에 발생하는 소화 파편의 크기로 제한됩니다. 그러나, 펩신으로 인해 분열의 비특이적 인 특성으로 생성 된 중복 조각은 공간 해상도의 증가로 이어질 수 있습니다. 또, 여러 다른 프로테아제는 급냉 조건 하에서 그러나 훨씬 덜 효율적인 열 펩신보다 활성 인 것으로 밝혀졌다. 또한 increa공간 해상도의 SE는 전자 캡쳐 해리 (ECD), 전자 전달 해리 (ETD) 및 적외선 다 광자 해리 (IRMPD) 11-13로서 중수소 패턴을 보존 방법에 의해 기체 상에있는 펩티드의 분열에 의해 도달 될 수있다. 이러한 기술에 의한 충돌 – 유도 해리 (CID)에 의해 관찰되는 분자 내 수소 이온 마이그레이션 ( "스크램블링"), 가장 일반적으로 사용되는 기술로 조각화 공간 해상도의 손실을 방지한다. 그러나 이러한 방법은 모든 개인 펩타이드에 대한 최적화를 필요로하고, 따라서 여전히 매우 도전이다.
HX-MS는 바이러스 캡시드 어셈블리 14-17 포함한 단백질 – 리간드와 단백질 – 단백질 상호 작용을 분석하는데 사용되어왔다. 단백질 전개 및 온도에 의한 구조적 변화가 7,18,19을 조사 하였다뿐만 아니라 접힘. 인산화 및 단일 아미노산 변이 관련 구조적는 16, 20 및 nucleot 변경IDE에 의한 변경 (21, 22)를 분석 하였다. 따라서,이 방법은 조립 및 분자 기계의 역학을 분석 할 이상적으로 적합한 것 같습니다. 그 메커니즘 위대한 장군 관심의 한 매력적인 후보,의 Hsp90 보호자 복잡합니다.
