O número de estruturas de cristal de proteínas e complexos de proteína aumentou rapidamente nos últimos anos. Apresentam instantâneos inestimáveis da organização estrutural das proteínas e fornecer uma base para a análise da estrutura-função. No entanto, a dinâmica das proteínas e as mudanças conformacionais, que são essenciais para as suas funções, são raramente revelado por cristalografia de raios-X. Cryo-electronmicroscopy, por outro lado, é capaz de capturar os complexos de proteínas e de proteínas em diferentes conformações, mas geralmente não é possível resolver as alterações conformacionais para baixo para o nível de uma estrutura secundária. Dinâmica de conformação de proteínas em solução em detalhes atómicas só pode ser resolvido por RMN, mas este método é ainda limitado a proteínas de tamanhos relativamente pequenos (geralmente ≤ 30 kDa) e necessita de altas concentrações de proteínas (≥ 100 pM), o que dificulta as experiências com oligomerização ou agregação de proteínas propensas 2. Um método queé capaz de ponte entre a alta resolução de cristalografia de raios-X e crio-electronmicroscopy e que não é limitado pelo tamanho ou a concentração de proteína é de hidrogénio amida-1 H / 2 H-troca (HX) em combinação com espectrometria de massa (MS). Nos últimos anos, este método tem desenvolvido para um instrumento analítico valioso para a análise da dinâmica das proteínas, o dobramento de proteínas, a estabilidade da proteína e alterações conformacionais 3-5. A base molecular deste método é a natureza lábil do backbone hidrogénios de amida em proteínas, que irá trocar com átomos de deutério, quando a proteína é colocada em um D 2 O solução. O consequente aumento da massa de proteína ao longo do tempo é medido com alta resolução MS.
Em péptidos não estruturados curtas só HX depende da temperatura, da concentração do catalisador (OH -, H 3 O + por exemplo pH, ver Figura 3) e as cadeias laterais de resíduos de aminoácidos adjacentes, devido à indutivo, gatoefeitos alytic e estéricos. Estes efeitos sobre o ch intrínseca taxa de permuta química k foram elegantemente quantificada por Bai et al. 6 e um programa está disponível (por cortesia Z. Zhang), que calcula k ch para cada aminoácido num polipéptido dependente do pH e da temperatura. Em pH neutro e temperatura ambiente k ch é da ordem de 10 -10 1 3 seg -1. Em proteínas dobradas HX pode ser 2-9 ordens de grandeza mais lento, principalmente devido a pontes de hidrogénio na estrutura secundária e a um grau menor devido ao acesso limitado de hidratadas OH – iões para o interior de uma proteína fortemente dobrado. HX em proteínas nativas, portanto, implica desdobramento, por permuta química parcial ou global e redobrando para o estado nativo de acordo com a equação (1) e as taxas de câmbio observadas k obs dependem da taxa de abertura op k, os cl k taxa de fechamento ea troca química intrínseca rate k ch de acordo com a equação (2).
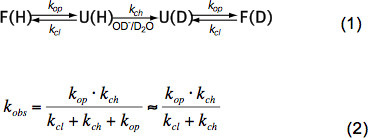
Sob condições de estado nativo k op é muito menor do k ch e pode ser negligenciada no denominador. Existem dois regimes cambiais extremos chamados EX1 e EX2. Se o cl k é muito menor do k ch (EX1) a taxa observada é praticamente igual à taxa de abertura e HX permite a observação imediata do desdobramento de um elemento estrutural. Tal regime cambial, onde todos os prótons amida troca de uma só vez aquando da abertura do elemento estrutural, é facilmente observáveis em MS por uma distribuição bimodal dos picos de isótopos 7. Se k cl é muito maior do que k ch (EX2) a taxa observada é proporcional à k ch em que a constante de proporcionalidade é igual aos equilíbrios-desdobramento dobráveis constante op K u = k </sub> K / cl. Sob estas condições, muitos eventos de abertura e fecho são necessárias antes de todos os protões amida troca de deutério, que conduz a um aumento gradual da massa média, enquanto a distribuição isotópica permanece praticamente a mesma. O regime EX2 permite a determinação da energia livre de desdobramento ÄG u e, por conseguinte, a estabilidade de um elemento estrutural. Sob condição de estado nativo do regime EX2 é mais comum. Aumento do pH e da adição de agentes caotrópicos pode mudar o mecanismo de troca de EX1. Portanto, HX-MS pode ser utilizada para explorar termodinâmico, bem como os parâmetros cinéticos de dobragem de proteína e alterações conformacionais.
Como mencionado acima HX é intrinsecamente pH e dependente da temperatura e meia-vida a troca de um protão exposta completamente solvente do grupo amida espinha dorsal é entre 5-400 ms a um pH fisiológico (pH 7,6) e 30 ° C, mas 10 min a> 15 horas, com uma média de> 2 horas a pH 2,9 e 0 °C (excepto para o protão da primeira estrutura de ligação amida de um polipeptídeo, que troca com uma meia-vida de aprox. 1-2 min). Sob tais condições lenta troca é possível para digerir a amostra usando proteases (por exemplo pepsina) que atuam nessas condições, sem perder todas as informações contidas nos dêuterons incorporadas. Desde a introdução de digestão péptica sob condições troca lenta, não apenas da cinética geral HX de proteínas de comprimento completo pode ser analisado, mas HX pode ser localizado em regiões específicas 8,9. A resolução espacial está limitado ao tamanho dos fragmentos gerados pépticas, que é, em geral, entre 10-30 resíduos. No entanto, fragmentos sobrepostos criados devido à natureza não específica da clivagem por pepsina pode levar a um aumento da resolução espacial. Além disso, várias outras proteases foram encontrados para ser activo em condições de têmpera, contudo, muito menos eficiente do que a pepsina 10. Além disso aumense de resolução espacial pode ser alcançado pela fragmentação de peptídeos em fase gasosa por meio de métodos que preservaram o padrão deuteração tais como a captura eletrônica de dissociação (ECD), transferência eletrônica de dissociação (ETD) e dissociação multiphoton infravermelho (IRMPD) 11-13. Estas técnicas de evitar a perda de resolução espacial devido à migração intramolecular de protões ("scrambling"), que é observada por dissociação induzida por colisão (CID) a técnica de fragmentação mais utilizada. No entanto, estes métodos requerem otimização para cada peptídeo individual e é, portanto, ainda bastante desafiador.
HX-MS foi utilizada para analisar interacções proteína-ligando e de proteína-proteína, incluindo a montagem da cápside viral 14-17. Proteína desdobramento e redobrando assim como temperatura provocadas mudanças conformacionais foram investigados 7,18,19. Fosforilação e conformacional relacionada com mutação único aminoácido muda 16,20 e nucleotmudanças ide-induzidas foram analisados 21,22. Portanto, esse método parece idealmente adequado para analisar a montagem e dinâmica de máquinas moleculares. Um candidato atraente, cujo mecanismo é de grande interesse geral, é o complexo chaperone Hsp90.
