Representative flow cytometry results of hMPC isolation from human muscle tissue can be viewed in Figure 1. hMPCs can be identified by first gating events based on side scatter and forward scatter to eliminate dead cells or debris, followed by selecting only cells which are negative for 7-AAD and therefore are viable. Selection of cells positive for both the cell surface markers CD56 and CD29 represents the hMPC population. A biopsy of 60 mg only provides approximately 75−250 hMPCs.
A representative confluence scan is shown in Figure 2A. The green outlines in Figure 2B were generated by the imaging cytometer and show how the imaging cytometer determined confluence based on the analysis settings selected. In both images shown, the confluence determined by the imaging cytometer agrees with the confluence visually determined by the user. However, these images also highlight that the imaging cytometer is not perfect. For example, the red arrow highlights an imperfection in the plate which is being counted as cells. If a large number of these imperfection are present on the plate, the resulting confluence will not accurately represent the confluence of the cells. Figure 2C shows a representation of all three channels used to count cells (brightfield, blue and red) and the merged image. Figure 2D shows heterogeneity between donors. Discovering and accurately characterizing this heterogeneity is a key application of the imaging cytometer. Figure 2E shows that confluence measurements and nuclei counts from unique donors are highly correlated.
Figure 3 provides a guideline for how to identify hMPCs based on the FACS procedure described in this protocol. Gating based on forward and side scatter allows for separation of viable cells from debris (Figure 3A). A comparison of forward scatter by height to forward scatter by area was used to distinguish events representing single cells (the boxed area in Figure 3B). Viable cells are marked by a lack of incorporation of the viability stain 7-AAD (Figure 3C). Finally, cells positive for both CD56 and CD29 are identified as hMPCs (Figure 3D). To validate the FACS procedure described in this protocol the selection of Pax7 positive cells can be determined by immunostaining cells with a Pax7-specific antibody and measuring expression on a flow cytometer. Figure 4 compares the number of hMPCs as determine by Pax7 immunostaining (Figure 4A) vs. the FACS procedure (Figure 4B) detailed in this protocol from the same population of cells. Figure 5 uses Pax7 immunostaining and analysis via flow cytometry to show the enrichment of Pax7 expressing cells in the total population after FACS (Figure 5A compared to Figure 5B) as well as the maintenance of the number of Pax7 expressing cells after passaging (Figure 5B compared to Figure 5C).
hMPCs can be differentiated to form myotubes by following section 6. To determine whether isolated hMPCs maintain myogenic capacity cells can be immunostained with an antibody specific for embryonic myosin heavy chain and visualized using fluorescent microscopy. Representative images of embryonic myosin heavy chain positive myotubes can be viewed in Figure 6.
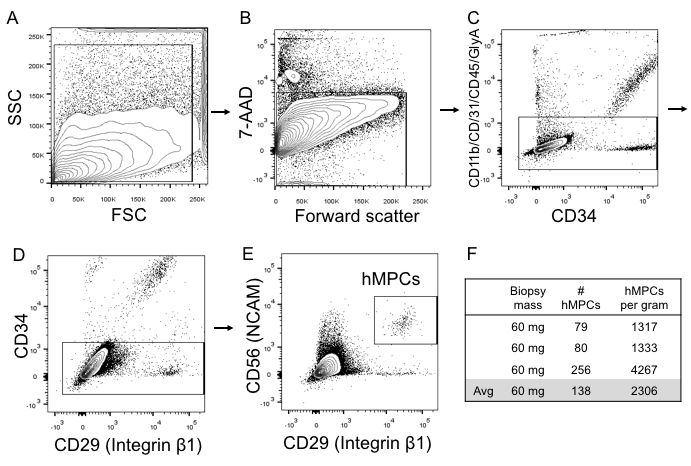
Figure 1: Representative flow cytometry images for hMPCs sorted directly out of muscle biopsy tissue. (A) Side scatter area (y-axis) vs. forward scatter area (x-axis) gating strategy used to identify all cells in a donor sample. (B) 7-AAD viability stain (y-axis) vs. forward scatter (x-axis) to identify live cells. (C) Negative selection sorting marker profile (-CD11b, -CD31, -CD45, -GlyA [y-axis]) vs. selection marker CD34. (D) Negative selection marker CD34 (y-axis) vs. the positive selection marker CD29. (E)Positive selection marker CD56 (y-axis) vs. the positive selection marker CD29 (x-axis). (F)Yields from three different skeletal muscle biopsies. FSC = forward scatter; SSC = side scatter; hMPCs, human muscle progenitor cells.
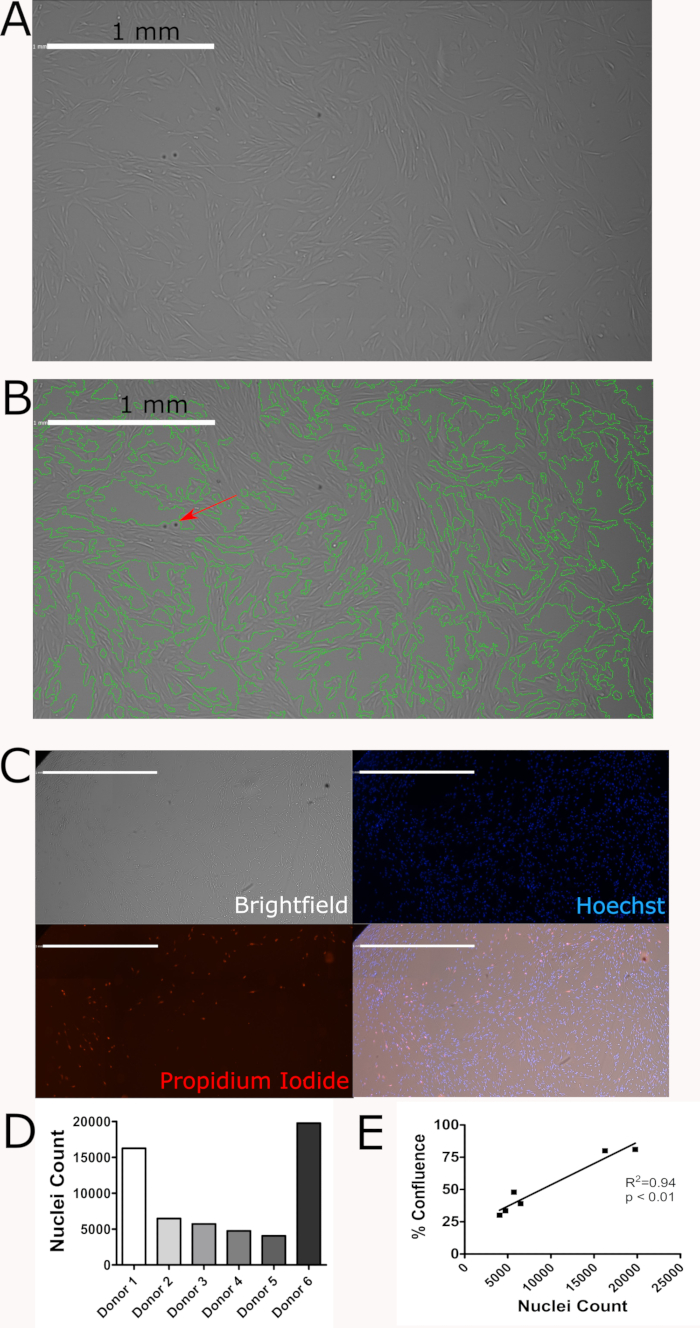
Figure 2: Proliferative potential of hMPCs derived from human donors is maintained after 6 passages. (A)Representative confluence scan. (B) Representative confluence scan with green outlines showing how the imaging cytometer determined confluence. The red arrow shows an imperfection on the plate. (C) Representative brightfield, Hoechst 33342 (blue), and propidium Iodide (red) staining and a merged image of these three channels. Scale bars = 1mm. (D) Nuclei counts from 6 unique donors highlights hMPC heterogeneity. (E) Confluence and nuclei count are highly correlated. Please click here to view a larger version of this figure.
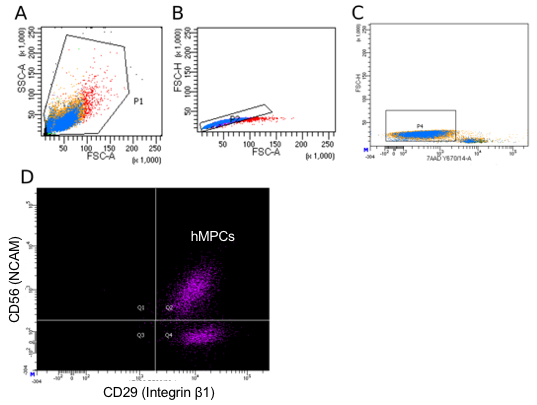
Figure 3: Representative flow cytometry sorting parameters for hMPC isolation. (A) Side scatter area (y-axis) vs. forward scatter area (x-axis) gating strategy used to identify all cells in a donor sample. (B) Forward scatter height (y-axis) vs. forward scatter area (x-axis) to disqualify doublets from the sorting population. (C) Forward scatter height (y-axis) vs. 7-AAD incorporation to identify viable cells. (D) PE-Cy7 (CD56) vs. AF488 (CD29) staining with Q2 representing double positive cells (hMPCs). Color represents the density of events.
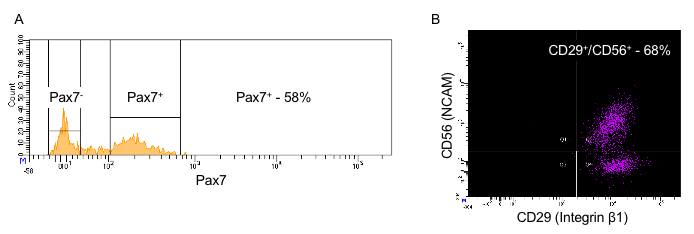
Figure 4: Comparison of CD29/CD56 positivity vs. Pax7 positivity in passage 4 hMPCs from the same donor. (A) Count (y-axis) vs. Pax7 expression (x-axis). (B)Positive selection marker CD56 (y-axis) vs. positive selection marker CD29 (x-axis). NCAM = neural cell adhesion molecule; hMPC = human muscle progenitor cell.
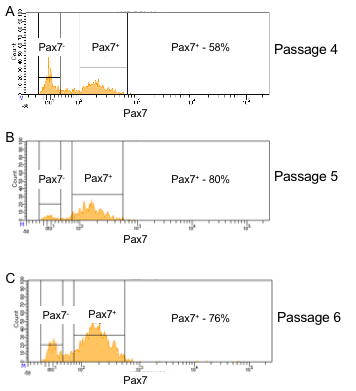
Figure 5: Pax7 positivity is maintained in hMPCs derived from the same donor over multiple passages. (A) Pax7 expression (x-axis) of cells which had been passaged 4 times prior to FACS sorting. (B) Pax7 expression (x-axis) of hMPCs 1 passage after FACS sorting for CD29 and CD56 positivity (5 total passages). (C) Pax7 expression (x-axis) of hMPCs 2 passages after FACS sorting for CD29 and CD56 positivity (6 total passages).
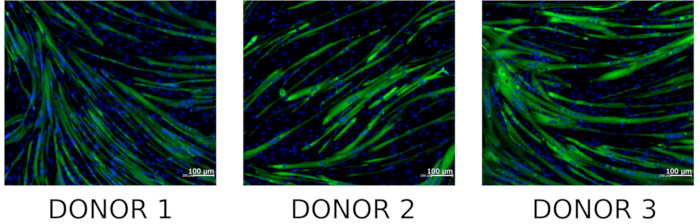
Figure 6: Staining of hMPC derived myotubes for embryonic myosin heavy chain. Representative microscopic images of differentiated hMPCs co-stained with DNA stain (Hoechst 33342, blue) and embryonic myosin heavy chain (green) (n = 3). Please click here to view a larger version of this figure.
