Purification of Ubiquitinated p53 Proteins from Mammalian Cells
Summary
The protocol describes a step-by-step method to purify ubiquitinated proteins from mammalian cells using the p53 tumor suppressor protein as an example. Ubiquitinated p53 proteins were purified from cells under stringent nondenaturing and denaturing conditions.
Abstract
Ubiquitination is a type of posttranslational modification that regulates not only the stability but also the localization and function of a substrate protein. The ubiquitination process occurs intracellularly in eukaryotes and regulates almost all basic cellular biological processes. Purification of ubiquitinated proteins aids the investigation of the role of ubiquitination in controlling the function of substrate proteins. Here, a step-by-step procedure to purify ubiquitinated proteins in mammalian cells is described with the p53 tumor suppressor protein as an example. Ubiquitinated p53 proteins were purified under stringent nondenaturing and denaturing conditions. Total cellular Flag-tagged p53 protein was purified with anti-Flag antibody-conjugated agarose under nondenaturing conditions. Alternatively, total cellular His-tagged ubiquitinated protein was purified using nickel-charged resin under denaturing conditions. Ubiquitinated p53 proteins in the eluates were successfully detected with specific antibodies. Using this procedure, the ubiquitinated forms of a given protein can be efficiently purified from mammalian cells, facilitating studies on the roles of ubiquitination in regulating protein function.
Introduction
Ubiquitin is an evolutionarily conserved protein of 76 amino acids1,2,3. Ubiquitin covalently binds lysine residues on target proteins through cascades involving activating (E1), conjugating (E2), and ligase (E3) enzymes. Ubiquitin is first activated by the E1 enzyme and is then transferred to the E2 conjugating enzymes. Subsequently, E3 ubiquitin ligases interact with both ubiquitin-loaded E2 enzymes and substrate proteins and mediate the formation of an isopeptide bond between the C-terminal of ubiquitin and a lysine residue in the substrate1,2,3,4,5. Ubiquitination involves the attachment of ubiquitin moieties to lysine residues on substrate proteins or to itself, leading to protein monoubiquitination or polyubiquitination. This ubiquitination process occurs intracellularly in eukaryotes and regulates a large variety of biological processes. Ubiquitination results in the degradation of substrate proteins via the ubiquitin-proteasome system1,2,3,4,5. In addition, ubiquitination modulates protein subcellular localization, protein complex formation, and protein trafficking in cells3,5. Ubiquitin moieties ligated to substrate proteins can be removed by deubiquitinating enzymes (DUBs)6,7. Notably, the different ways in which ubiquitin chains are assembled provide a myriad of means to regulate various biological processes1,5. The exact roles of ubiquitination in regulating substrate protein function remain incompletely understood till now. The purification of ubiquitinated proteins contributes to the elucidation of the effects of protein ubiquitination on a variety of cellular processes.
The p53 protein is one of the most important tumor suppressor proteins and exhibits genetic mutations or inactivation in almost all human cancers8,9,10,11. p53 stability and activity are delicately regulated in vivo by posttranslational modifications, including ubiquitination, phosphorylation, acetylation, and methylation12,13. The p53 protein has a short half-life ranging from 6 min to 40 min in various cells, which results mainly from its polyubiquitination and subsequent proteasomal degradation10,12. Mouse double minute 2 (Mdm2) is an E3 ubiquitin ligase of p53 that binds to the N-terminus of p53 to inhibit its transcriptional activity12,14,15. Mdm2 promotes the polyubiquitination and proteasomal degradation of p53 to control its stability and induces monoubiquitination of p53 to facilitate its nuclear export12,14,15,16. Here, Mdm2-mediated p53 ubiquitination is used as an example to introduce a method for the purification of ubiquitinated proteins from mammalian cells in detail. The regulators that influence the ubiquitination status of target proteins can be identified using this in vivo ubiquitination assay when they are overexpressed or knocked down/knocked out in mammalian cells. In addition, ubiquitinated proteins can be used as substrates for in vitro deubiquitination assay. A high-throughput screening can be performed to identify specific DUBs for target proteins by incubating ubiquitinated substrates with individual DUBs. Ubiquitinated proteins may act as a scaffold to recruit downstream signaling proteins in cells. A ubiquitinated target protein complex can be purified by sequential immunoprecipitation under native purification conditions and identified by mass-spectrometry. The current protocol can be extensively used to investigate the cellular proteins regulated by ubiquitination.
Several methods have been established to purify ubiquitinated proteins, which include the use of affinity-tagged ubiquitin, ubiquitin antibodies, ubiquitin-binding proteins, and isolated ubiquitin-binding domains (UBDs)17. Here, we provide a protocol using affinity-tagged ubiquitin as a mediator to purify ubiquitinated proteins in mammalian cells. The use of poly-His-tagged ubiquitin offers advantages over the other methods. Ubiquitinated proteins are purified in the presence of strong denaturants, which reduces non-specific binding to nickel-charged resin by linearizing cellular proteins and disrupting protein-protein interactions. In contrast, the use of ubiquitin antibodies, ubiquitin-binding proteins, and isolated UBDs as mediators cannot effectively exclude binding partners from target protein because purification needs to be performed under less stringent conditions. Moreover, purification may also lead to increased binding of unrelated proteins using these mediators. In addition, there is a binding propensity for various ubiquitin linkage types as well as mono- and poly-ubiquitination by ubiquitin-binding proteins or isolated UBDs17. The use of poly-His-tagged ubiquitin contributes to pull down all cellular ubiquitinated proteins. Alternatively, the use of commercially available anti-Flag or anti-HA antibody-conjugated agarose make it easier to immunoprecipitate large-scale Flag- or HA-tagged target proteins under nondenaturing conditions. A second purification step, for example, by nickel-charged resin targeting poly-His-tagged ubiquitin, can be used to acquire ubiquitinated target proteins with a high purity for downstream experiments. Notably, an epitope tagging purification strategy can be adapted when a specific antibody cannot be acquired to immunoprecipitate target proteins effectively. Finally, purification of ubiquitinated proteins in mammalian cells, in comparison with purification in vitro, retains the ubiquitin linkage mode of target proteins under more physiological conditions.
Protocol
NOTE: H1299 cells were kindly provided by the Stem Cell Bank, Chinese Academy of Sciences and were proven to be negative for mycoplasma contamination.
1. Cell culture
- For the initial culture, place 1 x 106 cells of human lung adenocarcinoma cell line, H1299 in a 10 cm Petri dish with 10-12 mL of RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 1% glutamine additive, 1% sodium pyruvate, and antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin). Maintain the cells at 37 °C in a humidified incubator with 5% CO2. Split the cells every 2-3 days depending upon when they reach 80%-90% confluency.
- One day before transfection, prepare 6-9 x 106 cells for three Petri dishes and plate 2-3 x 106 cells in a single 10 cm Petri dish containing 10-12 mL of medium to achieve a confluence of 70%-90% during transfection.
NOTE: HEK293T cells can also be used to purify ubiquitinated proteins due to their high transfection efficiency and protein expression level.
2. Plasmid transfection
- Add 1.5 mL of reduced serum medium in three 15 mL centrifuge tubes.
- Add plasmid DNA ready for transfection to each tube to make dilutions as follows. Tube 1: 1 µg of Flag-p53 plasmid, 12 µg of empty vector; Tube 2: 1 µg of Flag-p53 plasmid, 3 µg of His-HA-Ub (HH-Ub) plasmid, and 9 µg of empty vector; Tube 3: 1 µg of Flag-p53 plasmid, 3 µg of HH-Ub plasmid, and 9 µg of Mdm2 plasmid. Add a total of 0.2 µg green fluorescent protein (GFP) plasmid to each tube to monitor the transfection efficiency.
NOTE: A pilot experiment should be performed to determine the optimal amounts of plasmids needed to achieve efficient target protein expression in cells. - Add 78 µL of liposome transfection reagent to 4.5 mL of reduced serum medium in another centrifuge tube to dilute liposomes. Mix thoroughly by flicking the tube and allow to stand for 5 min at room temperature (RT).
- Add 1.5 mL of the diluted liposome solution into each tube from step 2.2 containing 1.5 mL of the diluted DNA solution. Mix thoroughly and crosslink the plasmids with the liposomes for at least 20 min at RT. Use a plasmid DNA: liposome ratio of 1:2 (µg:µL) for each transfection.
- Discard the original medium from the Petri dishes and add 9 mL of reduced serum medium into each dish. Add 3 mL of the liposome-DNA mixture to each dish and mix the solution by gently shaking the plate back and forth 3x, and then left and right 3x for even distribution of the mixture in the plate.
- Culture the cells in a humidified incubator at 37 °C with 5% CO2. Replace the medium after 4-6 h and continue to culture the cells for 24-36 h.
3. Cell collection
- After 24-36 h, treat the cells with MG132 at a final concentration of 10 µM for 4-6 h.
NOTE: MG132 is a peptide aldehyde that efficiently blocks the proteolytic activity of the 26S proteasome complex. The amounts of proteins ubiquitinated with lysine-48 (K48)-linked polyubiquitin chains can be increased after cells are treated with MG132 or other proteasome inhibitors. - Discard the medium, wash the cells 2x (taking care to not flush out the cells) with ice-cold phosphate buffered saline (PBS), and aspirate the cells with serological pipets.
- Add 1 mL of PBS to the dish, remove the cells by scraping with a clean scraper, and transfer the cell suspension into microcentrifuge tubes. Centrifuge at 700 x g for 5 min to collect cell pellets.
4. Purification of ubiquitinated proteins under nondenaturing conditions
- Prepare Flag lysis buffer (50 mM Tris-HCl (pH 7.9), 137 mM NaCl, 10 mM NaF, 1 mM ethylene diamine tetraacetic acid (EDTA), 1% Triton X-100, 0.2% sarkosyl, and 10% glycerol) freshly supplemented with protease inhibitor cocktail before use.
- Add 800 µL of ice-cold Flag lysis buffer to the cells in each tube, mix the cells using a vortex oscillator or pipette gun, and then incubate the mixture on a rotator at 4 °C for 30 min.
- Subject the mixture to 5-10 brief pulses of ultrasonication. Perform the ultrasonication on the ice at a sonication frequency of 20 kHZ and 80% amplitude with each pulse lasting for 1 s. Incubate the mixture on a rotator at 40 revolutions per minute (rpm) at 4 °C for 30 min.
- Centrifuge the ultrasonicated samples at 8,000 x g for 20-30 min at 4 °C. Transfer the supernatant to a new microcentrifuge tube.
- Aliquot 80 µL (1/10) of the cell extract and mix with 20 µL of 5x sodium dodecyl sulfate (SDS) loading buffer. Boil the samples at 98 °C for 5 min, cool on ice for 2 min, and store at -20 °C until use. Use these samples as the input group to monitor protein expression.
- Add 30 µL of anti-Flag M2 antibody-conjugated (Flag/M2) beads to the remaining cell extracts and incubate on a rotator at 4 °C for at least 4 h or overnight.
- Centrifuge at 1,500 x g for 2 min at 4 °C to collect the beads. Add 1 mL of ice-cold Flag lysis buffer to the beads and mix by inverting the tube several times.
- Centrifuge at 1,500 x g for 2 min at 4 °C to collect the beads. Repeat step 4.7 for 4-6 times.
- Add 40 µL of Flag peptides at a final concentration of 200 ng/µL to the beads and incubate on a rotator at 4 °C for 2 h to elute the bound proteins.
- Centrifuge at 1,500 x g for 5 min at 4 °C. Transfer the supernatant to a new microcentrifuge tube.
- Add 10 µL of 5x SDS loading buffer, boil the mixture at 98 °C for 5 min, cool on ice for 2 min, and store at -20 °C for Western blotting. Alternatively, store the eluate from step 4.10 directly at -80 °C for other downstream experiments.
NOTE: The amount of purified ubiquitinated proteins can be scaled up by increasing the number of cells and amounts of plasmids transfected. An epitope tagging strategy can be adapted to purify cellular complexes that bind specifically to ubiquitinated p53 under native purification conditions. By co-expressing Flag-p53, mdm2, and HA-ubiquitin, the ubiquitinated p53 protein complex may be immunoprecipitated by sequential Flag and HA antibody-conjugated agarose from mammalian cells. To keep binding partners of ubiquitinated p53 proteins, the less stringent BC100 lysis buffer (20 mM Tris-HCl (pH 7.9), 100 mM NaCl, 10% glycerol, 0.2 mM EDTA, 0.2% Triton X-100, and 1x protease inhibitor) should be used during the process of purification. The eluate from HA peptide is subjected to mass spectrometry to identify all partners that bind specifically to ubiquitinated p53.
5. Purification of ubiquitinated proteins under denaturing conditions
- Add 1 mL of ice-cold PBS to the cell pellets obtained in step 3.3 and mix evenly. Aliquot 100 µL (1/10 volume) of the cell suspension into a microcentrifuge tube as the input sample.
- Centrifuge at 700 x g for 5 min at 4 °C to collect the cell pellets. Add 80 µL of Flag lysis buffer to the input sample, mix the cells using a vortex oscillator or pipette gun, and lyse the cells on ice for 1 h.
- Centrifuge at 8,000 x g for 20-30 min at 4 °C. Aliquot 80 µL of the supernatant into a new microcentrifuge tube.
- Add 20 µL of 5x SDS loading buffer to the supernatant, boil at 98 °C for 5-10 min, cool on ice for 2 min, and store at -20 °C until use.
- Centrifuge the remaining 900 µL of the cell suspension at 700 x g for 5 min at 4 °C to collect the cell pellet. Add 1 mL of ubiquitin buffer 1 (UB buffer 1; 6 M guanidine-HCI, 0.1 M Na2HPO4, 6.8 mM NaH2PO4, 10 mM Tris-HCI (pH 8.0), and 0.2% Triton X-100, freshly supplemented with 10 mM β-mercaptoethanol and 5 mM imidazole) to the cell pellets and mix several times by pipetting up and down to evenly distribute the cells.
NOTE: The concentration of imidazole may vary from 5 mM to 20 mM to decrease nonspecific protein binding on the nickel-charged resin. Ubiquitin buffer contains guanidine hydrochloride, which inactivates both ligases and DUBs. β-mercaptoethanol is a clear, colorless liquid with a strong, unpleasant odor similar to rotten eggs. A high concentration solution will cause serious damage to the mucous membrane, upper respiratory tract, skin, and eyes. Wear gloves and goggles and operate in the fume hood. - Subject the cell lysates to 10-20 rounds of ultrasonication until the solution is no longer viscous. Perform the ultrasonication on the ice at a sonication frequency of 20 kHZ and 80% amplitude with each pulse lasting for 1 s. Centrifuge at 8,000 x g for 20-30 min at RT and transfer the supernatant to a new microcentrifuge tube.
- Add 30 µL of nickel-charged resin to the supernatant and incubate on a rotator set at 15 rpm for 4 h or overnight at RT. Centrifuge at 1,500 x g for 2 min at RT to collect the beads.
- Add 1 mL of UB buffer 1, incubate with rotation in a shaker for 10 min at RT, and centrifuge at 1,500 x g for 2 min at RT to collect the beads.
- Add 1 mL of ubiquitin buffer 2 (UB buffer 2; 8 M urea, 0.1 M Na2HPO4, 6.8 mM NaH2PO4, 10 mM Tris-HCI (pH 8.0), and 0.2% Triton X-100) to the beads, incubate with rotation in a shaker for 10 min at RT, and centrifuge at 1,500 x g for 2 min to collect the beads. Repeat this once more.
- To the beads, add 1 mL of PBS, incubate with rotation in a shaker for 10 min at RT, and centrifuge at 1,500 x g for 2 min at RT to collect the beads. Repeat this once more.
- Elute bound proteins by incubating the beads with 40 µL of imidazole at a final concentration of 0.5 M for 1 h at RT. Centrifuge at 1,500 x g for 2 min at RT.
- Transfer the supernatant to a clean microcentrifuge tube, add 10 µL of 5x SDS loading buffer, boil at 98 °C for 5 min, cool on ice for 2 min, and store at -20 °C for Western blotting. Alternatively, store the unprocessed eluate at -80 °C for other downstream experiments.
6. Detection of purified ubiquitinated proteins by Western blotting
- Resolve the samples from steps 4.11 and 5.12 by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and then transfer them to a nitrocellulose membrane by Western blotting to detect target proteins using the corresponding antibodies as described18.
- In brief, incubate the membrane by immersing in 20 mL of blocking solution containing 5% defat milk powder in TBST buffer (15 mM Tris-HCI; pH = 7.6, 4.6 mM Tris-base, 150 mM NaCI, freshly supplemented with 0.1% Tween-20 before use) at RT for 1 h. Then, incubate the membrane with primary antibodies at RT for 2 h or overnight at 4 °C. Use the following antibodies for each set. All primary antibodies were used at a dilution of 1:1000.
- Detect total p53 protein, including ubiquitinated p53, with an anti-p53 monoclonal antibody after immunoprecipitation with Flag/M2 agarose beads.
- Detect ubiquitinated p53 proteins with an anti-HA antibody or anti-ubiquitin antibody after immunoprecipitation with Flag/M2 agarose beads.
- Detect ubiquitinated p53 proteins with an anti-p53 monoclonal antibody after nickel-charged resin pulldown.
- Detect total ubiquitinated protein with an anti-HA antibody or anti-ubiquitin antibody after nickel-charged resin pulldown.
- Incubate the membrane with horseradish peroxidase (HRP)-conjugated secondary antibodies at RT for 1 h after washing 3x with TBST buffer for 5 min each. All secondary antibodies were used at a dilution of 1:3000. Then, wash the membrane with TBST buffer 3x for 5 min each with gentle agitation.
- Start chemiluminescence imaging system to detect signals from Western blotting. Prepare the substrate solution for HRP by mixing solutions A and B at a ratio of 1:1.
- Place the nitrocellulose membrane in the camera obscura face up and coat the substrate solution evenly on the membrane using a pipetting gun. Select the automatic exposure procedure to capture chemiluminescent signals and manually adjust the exposure time until an ideal signal is acquired.
Representative Results
The schematic diagram shows the Flag-tagged p53 (Flag-p53) and His/HA double-tagged ubiquitin (HH-Ub) proteins (Figure 1A). The procedures utilized to purify ubiquitinated proteins are summarized in Figure 1B. Poly-His-tagged ubiquitin can be ligated to target proteins in mammalian cells. Ubiquitinated proteins can be purified with Flag/M2 beads under nondenaturing conditions or by immobilized metal ion affinity chromatography (IMAC) under denaturing conditions (Figure 1B). Total p53 protein, including ubiquitinated p53, was immunoprecipitated with Flag/M2 beads from H1299 cells under nondenaturing conditions. The signal from ubiquitinated p53 protein, which appears as a smeared band from low to high molecular weight, was markedly increased when Mdm2 was ectopically expressed in these cells, suggesting that ubiquitinated p53 protein has been effectively purified from cells (Figure 2). Next, the cells were lysed under denaturing conditions, and total cellular ubiquitinated protein was pulled down with nickel-charged resin. Total cellular protein was detected by Western blotting using an anti-HA monoclonal antibody and ubiquitinated p53 protein was detected using an anti-p53 monoclonal antibody. The results showed that the total ubiquitinated protein level was unchanged but the ubiquitinated p53 protein level was dramatically increased after Mdm2 was overexpressed in these cells. These results indicated that total ubiquitin proteins containing ubiquitinated p53 were effectively pulled down from cell lysates under denaturing conditions (Figure 3).
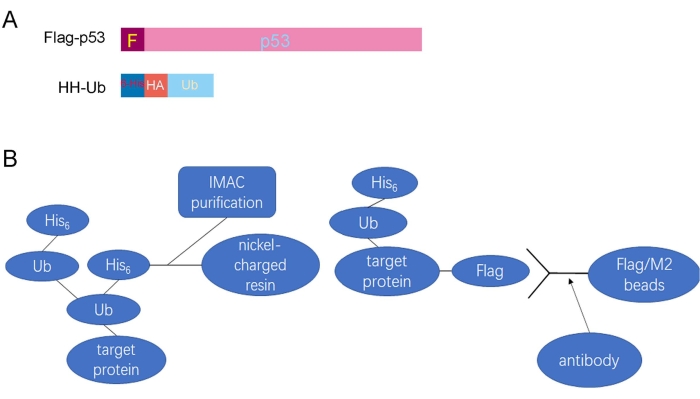
Figure 1: A summary of the procedures utilized to purify ubiquitinated proteins. (A) Schematic diagram of the Flag-tagged p53 (Flag-p53) and His/HA double-tagged ubiquitin (HH-Ub) proteins. (B) Ubiquitinated proteins can be purified with Flag/M2 beads under nondenaturing conditions and by IMAC under denaturing conditions. Abbreviations: His/HA = polyhistidine/hemagglutinin; Ub = ubiquitin; Flag/M2 beads = anti-Flag M2 antibody-conjugated beads; IMAC = immobilized metal ion affinity chromatography. Please click here to view a larger version of this figure.
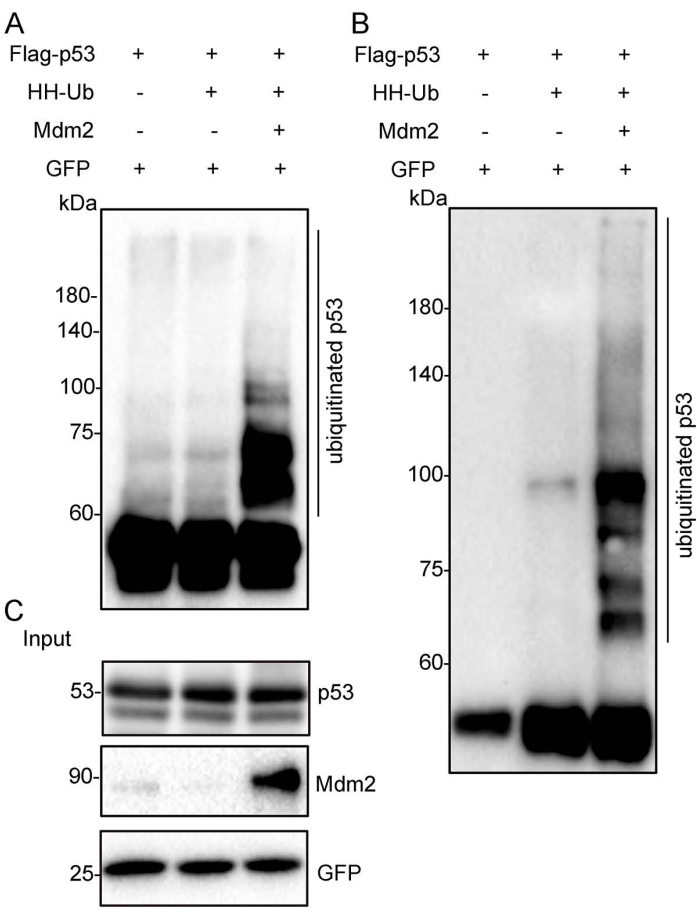
Figure 2: Ubiquitinated p53 proteins were purified under nondenaturing conditions. The Flag-p53 expression plasmid was transfected alone, with HH-Ub, or with Mdm2 and HH-Ub into H1299 cells. The total p53 proteins and ubiquitinated forms were immunoprecipitated by Flag/M2 beads from cell extracts. The eluate was subjected to Western blotting with anti-p53 (A) and anti-HA (B) monoclonal antibodies. (C) The crude cell extracts (Input) were subjected to Western blotting with anti-p53 and anti-Mdm2 monoclonal antibodies. GFP was used as the loading control. Abbreviations: HH-Ub = His/HA double-tagged ubiquitin; HA = hemagglutinin; Flag/M2 beads = anti-Flag M2 antibody-conjugated beads; GFP = green fluorescent protein. Please click here to view a larger version of this figure.
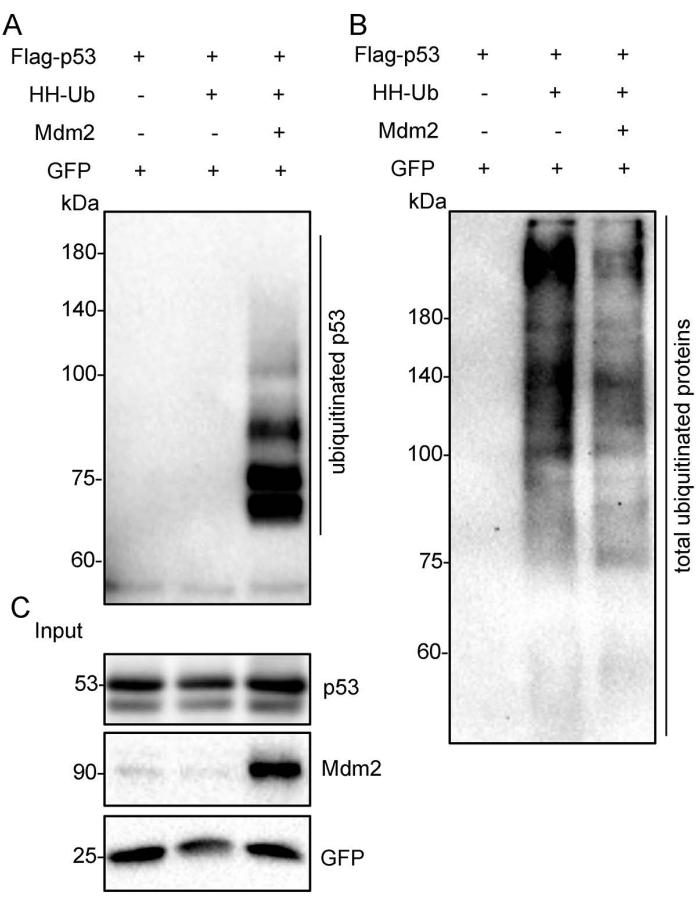
Figure 3: Ubiquitinated p53 proteins were purified under denaturing conditions. The Flag-p53 expression plasmid was transfected alone, with HH-Ub, or with Mdm2 and HH-Ub into H1299 cells. The total cellular ubiquitinated proteins were pulled down with nickel-charged resin and were then subjected to Western blotting with anti-p53 and anti-HA monoclonal antibodies. (A) Ubiquitinated p53 protein was detected using an anti-p53 antibody. (B) Total ubiquitinated protein was detected using an anti-HA antibody. (C) The crude cell extracts (Input) were subjected to Western blotting with anti-p53 and anti-Mdm2 monoclonal antibodies. GFP was used as the loading control. Abbreviations: HH-Ub = His/HA double-tagged ubiquitin; HA = hemagglutinin; GFP = green fluorescent protein. Please click here to view a larger version of this figure.
Discussion
Ubiquitination plays a critical role in almost all physiological and pathological cellular processes2. In recent years, great progress has been made in understanding the molecular role of ubiquitin in signaling pathways and how changes in the ubiquitin system lead to different human diseases2. The purification of ubiquitinated proteins contributes to providing insight into the exact roles of ubiquitination in these processes. The mixtures of ubiquitin-conjugated proteins can be purified from cells under nondenaturing and denaturing conditions. IMAC using nickel resins can be used to purify poly-His-tagged proteins under denaturing conditions. The goal of purifying ubiquitinated proteins under denaturing conditions is to discard the interaction partners from target proteins. All cellular components binding to the target proteins will be deleted from the elution under denaturing conditions. In contrast, some cellular proteins binding to target proteins may be kept in the elution under native conditions, which may interfere with the downstream experiments. Ubiquitinated proteins can be purified even in the presence of strong denaturants, which greatly decreases nonspecific protein binding to nickel resins. In contrast, the total target proteins, including their ubiquitinated forms, can be purified by epitope tagging strategies using antibody-conjugated agarose beads under nondenaturing conditions. Proteins were purified under less stringent conditions because antibody-mediated affinity purification was used during this process. The target proteins and their ubiquitinated forms were detected with protein- or ubiquitin-specific antibodies. HA- and Myc-tagged proteins can be conveniently purified using the same procedure with antibody-conjugated agarose beads. Notably, it is easier to detect proteins with single or multiple monoubiquitin chains than those with polyubiquitin chains with protein-specific antibodies. In contrast, proteins with polyubiquitin chains can be more easily recognized by ubiquitin-specific antibodies.
To effectively purify ubiquitinated proteins, the expression level of target proteins should be high in mammalian cells. Expression vectors with strong CMV promoters can be used to achieve high levels of protein expression in cells. In addition, E3 ubiquitin ligase and ubiquitin can be co-expressed with the target proteins. It will add ubiquitin moieties more effectively to the proteins. The activity of 26S proteasome can be blocked by small-molecule inhibitor in order to accumulate ubiquitinated proteins in cells. To make ubiquitinated proteins effectively bind to the resin, cell extracts should be briefly sonicated to disrupt the DNA complex to make the solution non-viscous. To decrease non-specific binding, a low concentration of imidazole should be added to the lysis buffer under denaturing conditions. The ubiquitinated proteins with very high molecular weight are easier to tether with the resin after the denaturing and refolding procedure. In order to increase the efficiencies of elution, try to use a higher concentration of imidazole. If the efficiencies of elution were still extremely low, try to elute the ubiquitinated proteins by directly boiling in SDS Laemmli loading buffer. Most of ubiquitinated proteins bound to the resin can be eluted in this strong denaturing buffer. To achieve a higher purity of ubiquitinated proteins, a two-step affinity purification can be adapted. After Flag/M2 immunoprecipitation under nondenaturing conditions, a second step purification by nickel-charged resin can be used to pull down His-tagged ubiquitinated protein and get rid of non-ubiquitinated substrates. In contrast, after nickel-charged resin pull-down under denaturing condition, a second affinity purification step can be adapted to immunoprecipitate the target proteins using the epitope tagging strategy by anti-Flag or anti-HA antibody conjugated agarose beads.
We have used more sensitive immunoblotting instead of silver staining or Coomassie staining to detect ubiquitinated proteins because we performed a small scale of purification in this study. In fact, although some cellular proteins are easily ubiquitinated in cells, only a small part of proteins can be ligated with ubiquitin moieties. To successfully detect these proteins by silver staining or Coomassie staining, a large scale of purification should be performed. To increase the purity of ubiquitinated proteins, a second affinity purification step should also be done. We did not perform these experiments because ubiquitinated proteins purified through this protocol completely meet the requirement of downstream analyses. For example, in order to identify specific DUBs for a target protein regulated by ubiquitination, we need to purify a large amount of ubiquitinated target proteins. These proteins can be incubated with individual DUBs purified from mammalian cells in a deubiquitination buffer. By this in vitro high throughput screening, we can identify DUBs specific for a target protein in mammalian cells.
There are several limitations of the technique. The ubiquitinated proteins account for a very small part of target proteins, it is difficult to purify a large amount of ubiquitinated target proteins. Try to scale up the protein expression in cells in order to achieve high purification efficiency. An alternative is to perform an in vitro ubiquitination reaction to acquire more ubiquitinated proteins. Some proteins are difficult to be highly expressed in the mammalian cells, which puts another limitation on purifying higher quantities of ubiquitinated proteins. Finally, the current protocol is limited to the purification of tagged proteins that are overexpressed using plasmid DNA. The expression levels of the target proteins are higher than that of the endogenous target proteins. To assess the endogenous ubiquitination status of a target protein, the proteins with their ubiquitinated forms can be directly immunoprecipitated by target protein-specific antibodies from cells. Anti-ubiquitin antibodies can be used to determine the ubiquitination status of the target protein. However, under some circumstances, the lower expression level, the lower ubiquitination level of the target protein, or the lower immunoprecipitation efficiency of specific antibodies may limit the detection of ubiquitination status of the endogenous target proteins from mammalian cells. The current protocol may provide an experimental system instead.
The p53 tumor suppressor protein plays a critical role in preventing the malignant transformation of normal cells8,9,10,11. The stability of p53 is tightly regulated by ubiquitination-mediated degradation in cells. Mdm2 acts as an E3 ubiquitin ligase to promote p53 mono- and polyubiquitination in a dose-dependent manner16. Here, ubiquitinated p53 proteins were purified under denaturing and nondenaturing conditions in mammalian cells in presence of Mdm2 overexpression. The monoubiquitinated p53 proteins were preferentially detected with anti-p53 monoclonal antibodies, whereas the polyubiquitinated forms were easily detected with ubiquitin-specific antibodies. The levels of polyubiquitinated forms can be effectively increased by elevating the expression of Mdm2 in cells. This paper provides an ideal experimental system for the preparation of ubiquitinated proteins in order to elucidate the role of ubiquitination in modulating protein function. Ubiquitinated proteins can be used to identify specific DUBs for a target protein and to reveal the roles of different types of ubiquitin chains, for example, the K48- and K63-linked chains, in signaling. This protocol can be used to investigate the roles of ubiquitination in protein-protein interaction and protein complex formation.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
This work was supported by a grant from the National Natural Science Foundation of China (81972624) to D.L.
Materials
| β-mercaptoethanol | Sangon Biotech | M6250 | |
| Amersham ECL Mouse IgG, HRP-linked whole Ab (from sheep) | GE healthcare | NA931 | Secondary antibdoy |
| Amersham ECL Rat IgG, HRP-linked whole Ab (from donkey) | GE healthcare | NA935 | Secondary antibdoy |
| Anti-Flag M2 Affinity Gel | Sigma-Aldrich | A2220 | FLAG/M2 beads |
| Anti-GFP monocolonal antibody | Santa cruz | sc-9996 | Primary antibody |
| Anti-HA High Affinity | Roche | 11867423001 | Primary antibody |
| Anti-Mdm2 monocolonal antibody (SMP14) | Santa cruz | sc-965 | Primary antibody |
| Anti-p53 monocolonal antibody (DO-1) | Santa cruz | sc-126 | Primary antibody |
| EDTA | Sigma-Alddich | E5134 | solvent |
| Fetal Bovine Serum | VivaCell | C04001-500 | FBS |
| FLAG Peptide | Sigma-Alddich | F3290 | Prepare elution buffer |
| GlutaMAX | Gibco | 35050-061 | supplement |
| Guanidine-HCI | Sangon Biotech | A100287-0500 | solvent |
| H1299 | Stem Cell Bank, Chinese Academy of Sciences | ||
| Image Lab | Bio-rad | software | |
| Immidazole | Sangon Biotech | A500529-0100 | solvent |
| Immobilon Western Chemiluminescent HRP Substrate | Millipore | WBKLS0500 | |
| Lipofectamine 2000 reagents | Invitrogen | 11668019 | Transfection reagent |
| Na2HPO4 | Sangon Biotech | A501727-0500 | solvent |
| NaCl | Sangon Biotech | A610476-0005 | solvent |
| NaF | Sigma-Alddich | 201154 | solvent |
| NaH2PO4 | Sangon Biotech | A501726-0500 | solvent |
| Ni-NTA Agarose | QIAGEN | 30230 | nickel-charged resin |
| Nitrocellulose Blotting membrane | GE healthcare | 10600002 | 0.45 µm pore size |
| Opti-MEM reduced serum medium | Gibco | 31985-070 | Transfection medium |
| PBS | Corning | 21-040-cv | |
| Penicillin-Streptomycin Solution | Sangon Biotech | E607011-0100 | antibiotic |
| Protease inhibitor cocktail | Sigma-Aldrich | P8340 | |
| RPMI 1640 | Biological Industries | 01-100-1ACS | medium |
| Sarkosyl | Sigma-Alddich | L5777 | solvent |
| SDS Loading Buffer | Beyotime | P0015L | |
| Sodium Pyruvate | Gibco | 11360-070 | supplement |
| Tris-base | Sangon Biotech | A501492-0005 | solvent |
| Tris-HCI | Sangon Biotech | A610103-0250 | solvent |
| Triton X-100 | Sangon Biotech | A110694-0500 | reagent |
| Tween-20 | Sangon Biotech | A100777-0500 | supplement |
| Ultra High Sensitive Chemiluminescence Imaging System | Bio-rad | ChemiDoc XRS+ | |
| Urea | Sangon Biotech | A510907-0500 | solvent |
Riferimenti
- Kwon, Y. T., Ciechanover, A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends in Biochemical Sciences. 42 (11), 873-886 (2017).
- Popovic, D., Vucic, D., Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nature Medicine. 20 (11), 1242-1253 (2014).
- Zheng, N., Shabek, N. Ubiquitin ligases: Structure, function, and regulation. Annual Review of Biochemistry. 86, 129-157 (2017).
- Dikic, I., Wakatsuki, S., Walters, K. J. Ubiquitin-binding domains – from structures to functions. Nature Reviews Molecular Cell Biology. 10 (10), 659-671 (2009).
- Oh, E., Akopian, D., Rape, M. Principles of ubiquitin-dependent signaling. Annual Review of Cell and Developmental Biology. 34, 137-162 (2018).
- Clague, M. J., Urbe, S., Komander, D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nature Reviews Molecular Cell Biology. 20 (6), 338-352 (2019).
- Harrigan, J. A., Jacq, X., Martin, N. M., Jackson, S. P. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nature Reviews Drug Discovery. 17 (1), 57-78 (2018).
- Vogelstein, B., Lane, D., Levine, A. J. Surfing the p53 network. Nature. 408 (6810), 307-310 (2000).
- Boutelle, A. M., Attardi, L. D. p53 and Tumor suppression: It takes a network. Trends In Cell Biology. 31 (4), 298-310 (2021).
- Levine, A. J. p53: 800 million years of evolution and 40 years of discovery. Nature Reviews Cancer. 20 (8), 471-480 (2020).
- Vousden, K. H., Prives, C. Blinded by the light: The growing complexity of p53. Cell. 137 (3), 413-431 (2009).
- Brooks, C. L., Gu, W. p53 ubiquitination: Mdm2 and beyond. Molecular Cell. 21 (3), 307-315 (2006).
- Liu, Y., Tavana, O., Gu, W. p53 modifications: exquisite decorations of the powerful guardian. Journal Of Molecular Cell Biology. 11 (7), 564-577 (2019).
- Dobbelstein, M., Levine, A. J. Mdm2: Open questions. Cancer Science. 111 (7), 2203-2211 (2020).
- Kulikov, R., et al. Mdm2 facilitates the association of p53 with the proteasome. Proceedings of the National Academy of Sciences of the United States of America. 107 (22), 10038-10043 (2010).
- Li, M., et al. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 302 (5652), 1972-1975 (2003).
- Tomlinson, E., Palaniyappan, N., Tooth, D., Layfield, R. Methods for the purification of ubiquitinated proteins. Proteomics. 7 (7), 1016-1022 (2007).
- Green, M., Sambrook, J. . Molecular Cloning A Laboratory Manual. (Fourth Edition). 2, 28 (2012).
