1. Extraction of RNA from large volumes of plasma
An overview of the single copy assay (SCA) and the single genome sequencing (SGS) protocol are provided in Figures 1 and 2.
- To obtain 7 ml of plasma, spin approximately 14 ml of blood collected in 15 ml EDTA (not heparin) collection tubes at 2,600 xg for 15 minutes without braking. Pipette plasma (making sure to avoid the buffy coat layer) into 15 ml tubes.
- If RNA is being extracted for the single copy assay (SCA), spike the plasma with 30,000 copies of the Rous Sarcoma Virus internal virion control (RCAS). The RCAS virus is prepared prior to performing SCA as described previously (20). Briefly, RCAS virus from cell culture supernatant is quantified by RT activity, diluted to a final concentration of 30,000 virions per 200 ul, diluted in RPMI tissue culture media with 5% FBS, and stored at -80°C in single use aliquots. The RCAS virus should not be frozen/thawed prior to use in the assay. RCAS is spiked into the patient plasma to control for the efficiency of the RNA extraction and for the accuracy of PCR quantification. More information about RCAS can be found at the following link: http://home.ncifcrf.gov/hivdrp/RCAS/.
- “Pre-spin” plasma, in 15 ml conical centrifuge tubes, at 2,600 xg for 15 minutes to separate out any lipids and cellular debris, which can interfere with accurate RNA quantification.
- Label Seton Easy-Seal centrifuge tubes with a permanent marker to identify sample number and the location where the pellet will form in the tube. After the pre-spin, transfer plasma to a Seton Easy-Seal centrifuge tubes by pipetting the plasma and avoiding any cellular debris and/or lipids at the surface. Record the input volume of plasma.
- Add Tris-buffered saline (TBS) to fill remaining volume of the Seton-Easy Seal tube to the bottom of the threaded neck. Be sure that NO BUBBLES are present in the tube or the tube will collapse in the ultracentrifuge. Make sure that the mark on the tubes is facing outward when placing the tubes into the rotor.
- Seal with re-usable caps and place samples in a pre-cooled Sorval T1270 rotor and centrifuge in the Sorval 90SE ultracentrifuge at 170,000 xg (50,000 rpm) at 4°C for 30 min.
- After centrifugation remove supernatant and add 90 ul molecular grade water and 10 ul proteinase-K (20 mg/ml) to the viral pellet (this will not be visible).
- Place tubes in a 55°C water bath and incubate for 30 minutes. Make sure the tubes are set at an angle so that the side with the pellet is immersed in the proteinase-K and water mixture.
- After incubation, shortly spin the tube in a table top centrifuge to collect all liquid at the bottom of the tube (about 5 seconds) and add 315 ul of 6M guanidinium thiocyanate solution and 10 ul of 20 mg/ml glycogen (Note: Follow appropriate disposal guidelines for this hazardous material; do not mix with bleach or acids and dispose in separate container).
- Vortex lightly and spin shortly (for about 5 seconds). Incubate for 10 minutes at room temperature and then transfer contents of each tube to a newly labeled 1.5 ml RNAse Free centrifuge tubes.
- Add 495 ul of molecular grade isopropyl alcohol to each tube, vortex for 10 seconds and centrifuge at 21,000 xg for 30 minutes at room temperature.
- Discard supernatant and add 500 ul of 70% ethanol.
- Vortex for 10 seconds and centrifuge at 21,000 xg for 15 minutes at room temperature. Remove ethanol with a transfer pipette. Spin again and remove any residual ethanol with a pipette. Air dry for 10 minutes. (For the SGS protocol dissolve RNA in 40 ul of 5 mM Tris-HCl pH 8.0,and continue with the SGS protocol).
- Dissolve pellet in 55 ul of RNA-buffer. RNA-buffer is obtained by adding 10 ul of 100 mM DTT and 25 ul of 40 U/ul Rnasin to 965 ul Tris-HCl (pH 8.0, 5 mM). Place on ice.
2. The Single Copy assay
A 96-well plate holds 8 patient samples including both HIV and RCAS standards and controls. This protocol will describe the set-up of a single plate.
- Start by preparing 2 aliquots of 1 ml RNA-buffer in which the HIV-1 RNA and RCAS RNA transcripts will be diluted for the RNA standard curves. RNA-buffer is described under 1.13.
- Prepare dilutions on ice in 2 ml centrifuge tubes. Make half-log dilutions of HIV-1 RNA transcripts in RNA-buffer by adding 25 ul of the HIV-1 RNA stock (1×106 copies/ul) to 54 ul RNA-buffer giving 3×105 copies/ul.
- Continue by making consecutive half-log dilutions (25 ul + 54 ul) down to 0.3 copies per 10 ul (Note: 0.3, 1, and 3 dilutions will not be a part of the standard curve during analysis for these are used as extrapolation values and should be set to unknowns during analysis). RCAS transcripts are stocked at 1×106 copies/ul. Similarly, prepare half-log dilutions of RCAS transcripts in RNA-buffer but down to 100 copies per 10 ul.
- Prepare the RT cocktail for the cDNA synthesis as listed in Table 1. Prepare RT and NRT reactions as listed in Table 1 keeping in mind to make up a little extra to account for any loss that may occur during transfer. Note: for the NRT cocktail, the RT enzyme is replaced by water to control for amplification from DNA.
Note: This step has recently been automated in addition to plate set up on a Qiagen Robot (originally Corbett). - Set up the optical 96-well plate (as shown in Figure 3), by adding 20 ul of RT reaction mixture to each well. In the 8 wells labeled “NRT,” add the cocktail without the reverse transcriptase (NRT reaction mixture). After adding the cocktails to the plate, add 10 ul of water to wells labeled “No template controls (NTC)”. Add 10 ul of HIV transcripts to wells labeled “HIV” in concentrations shown in Figure 3. Add 10 ul of RCAS transcripts to wells labeled “RCAS” in according concentrations. Add 10 ul of patient sample in the 3 adjacent wells labeled “Sample” on plate diagram. Add 10 ul of the eluted sample to the wells labeled “NRT.” Add 5 ul of the same sample and 5 ul of water to the wells labeled “internal control.”
- Seal the plate and run on thermocycler at: 25°C for 15 minutes, 42°C for 40 minutes, 85°C for 10 minutes, 25°C for 30 minutes, and 5°C hold.
- Prepare PCR master mixes according to Table 2.
- When the cDNA synthesis is completed, move to an area designated for using cDNA. In this designated area, add 20 ul of the PCR master mix to each well of the cDNA plate for both HIV and RCAS. It is important to perform this step in a designated area to avoid any possible contamination.
- Spin plate, seal with ABI or Roche Optical cover and cover with optical blanket (blanket only necessary for ABI 7700). Run on Roche 480 or ABI 7700. PCR Conditions: 95°C for 10 minutes, then 45 cycles of 95°C for 15 seconds, 60°C for 1 min. Separate analysis is required for RCAS and HIV. Examples of outcomes are shown in Figure 4. The slope of the standard curve will be affected by a normal Poisson distribution of low copy number transcripts. In order to ensure the most accurate slope for the standard curve, these low copy number standards (0.3, 1, and 3 copies) are set as “unknowns.”17
3. Single Genome Sequencing
- cDNA synthesis. Add 5 ul of 10 mM dNTPs and 5 ul of the 2 mM gene-specific primer (pol or env) to a well in a 96 well PCR plate (Biorad). Add 40 ul of the extracted RNA. Seal plate.
- Denature the RNA-mixture in thermocycler at 65°C for 10 min.
- Mix the reagents for cDNA synthesis, listed in Table 3. After the denaturing step, place PCR plate on ice, add the 50 ul of the cDNA mixture to each sample.
- Run PCR plate on thermocycler: 45°C for 50 min, 85°C for 10 min and 4°C hold.
- First PCR, 10 ul reactions. Prepare the PCR reaction mixture in a reagent tray using either the primers for first round amplification of p6-rt or env (Reagents listed in Table 3, primers listed in Table 5).
- Using a multi-channel pipette dispense 8.0 ul of PCR mixture to 73 wells on the 96 well PCR plate (70 samples and 3 negative controls).
- After cDNA synthesis is completed move the cDNA and PCR plate containing PCR reaction mix to an area designated for using cDNA. In the designated area, add 40 ul of Tris-HCl pH 8.0 to each cDNA sample bringing the final volume to 140 ul. Add 2.0 ul of cDNA to each of the 70 wells and 2.0 ul of water to each of the negative controls. Seal PCR plate. Run on thermocycler, program in Table 4.
- Nested PCR – 20ul reactions. Mix the reagents for the nested PCR in the reagent tray (reagents listed in Table 3, primers listed in Table 5).
- Add 18 ul of nested PCR mixture to 73 wells on a 96 well PCR plate, by multichannel pipette
- Dilute first PCR 1:5 and add 2 ul of first round PCR to the nested PCR. Run PCR reaction on the thermocycler, under the conditions listed in Table 4.
- Run samples on a 1% agarose gel. To ensure that the majority of the PCR products are the result of a single molecule of cDNA, no more than 30% of the wells should be positive. The process has been automated using the Beckman Coulter Biomek FM robot to load the contents of PCR plates to a 1%, 96 wells E-gel (Invitrogen). E-gels are run for 7 minutes on the E-base. Sequence products using primers listed in Table 5.
4. Representative results:
SCA:
All the controls should pass in order to consider the HIV-1 RNA measurement correct. If a negative control is positive, the run should be disregarded because of possible contamination. In order to control for losses of RNA during the extraction step, at least 50% of the RCAS spiked in plasma should be recovered. If the average RCAS is <15,000 copies, the sample fails and should be disregarded because a significant amount of RNA could have been lost during extraction or other steps of the protocol. This occurs in about 10% of all patient samples tested likely due to high lipids in plasma6. The recovery of the RCAS virus is meant to control for the quality of the extraction and the efficiency of cDNA synthesis and PCR amplification. It is not used to correct for HIV recovery since HIV is likely bound to immunoglobulins and other human proteins making it slightly different from virus isolated from tissue culture. The recovery of the RCAS in our lab was on average 25,716 +/- 4,057 copies/reaction mixture, or about 95% +/-15% of the RCAS RNA added17. The NRT (no reverse transcriptase) control is run in parallel in order to test for the amplification from DNA. The NRT value is subtracted from each of the three HIV-1 RNA values, then the average is calculated and if this number is less than zero (the amplification of DNA exceeds the amplification of RNA), the result would fail and should be disregarded because of risk of amplification from DNA rather than RNA. If HIV-1 RNA is only amplified from 1/3 wells, re-testing the sample is recommended to ensure that the amplification is true for the actual sample being assayed and not due to a possible contaminant.
If all the controls pass, the average HIV-1 RNA copy number in plasma can be calculated.
Example 1: If the average HIV-1 copy number is zero, the limit of detection is calculated based on the volume of plasma assayed. As an example; let’s say 7 ml of plasma was assayed. The smallest amount of RNA that could have been detected with this assay would have been 1 copy in one of the wells and 0 copies in the two other wells giving an average of 0.33 copies per well. The average copy number then has to be multiplied by a factor of 5.5 to get to the total copy number in the RNA elution (there is 10 ul in each well, but the total elution volume was 55 ul). The lower limit of detection is then: 5.5 x 0.33 copies divided by 7ml = 1.83/7=0.3 copies/ml. The concentration of HIV-1 RNA in plasma in this example was<0.3 copies/ml.
Example 2: HIV-1 RNA is detectable. RNA was extracted from 7 ml of plasma. The average copy number of HIV-1 RNA per well is calculated to be 2.0 copies per well. Then the average copy number per ml of plasma is 5.5 x 2.0 copies/7 ml = 1.6 HIV-1 RNA/ml of plasma.
A template Excel sheet used to calculate copy numbers can be down loaded from the website.
SGS:
If one of the negative controls is positive the run should be disregarded due to possible contamination. The number of product from each run will depend on the viral load in each sample and the condition of the sample. In our experience a lot of lipids or cells in plasma will reduce the chance of obtaining product. Storing conditions and previous freezing and thawing of the sample will also influence the recovery of RNA greatly. In general, when working with samples with viral loads below 50 copies/ml, product (bands on gel) is to be expected in about 1/3 -1/2 of the samples processed. Depending on the above mentioned factors, 1-5 bands per plate should be considered a good result due to the low viral loads in these patients.
| cDNA synthesis (RT Cocktail/cDNA reaction) | 1 reaction | No reverse transcriptase (NRT) cocktail/cDNA reaction (1 reaction) |
| Molecular Grade H2O | 8.1 ul | 8.2 ul |
| 25 mM MgCl2 | 6 ul | 6 ul |
| 25 mM dNTPs | 0.6 ul | 0.6 ul |
| 100 mM DTT | 0.2 ul | 0.2 ul |
| Random Hexamers (0.1 ug/ul) | 1.5 ul | 1.5 ul |
| 10X TaqMan Buffer A | 3.0 ul | 3.0 ul |
| Rnasin (40 U/uL) | 0.5 ul | 0.5 ul |
| Strategene RT (200 U/ul) | 0.1 ul | Do not add |
| Total | 20 ul | 20 ul |
| Sample RNA | 10 ul | 10 ul |
| Total volume | 30 ul | 30 ul |
Table 1. Reaction mixtures for cDNA synthesis in Single Copy Assay.
| PCR master mix | 1 reaction | Primers |
| H2O | 15.1 ul | HIV Forward Primer 5′-CATGTTTTCAGCATTATCAGAAGGA-3′ |
| 10X PCR Gold Buffer | 2.0 ul | HIV Reverse Primer 5′-TGCTTGATGTCCCCCCACT-3′ |
| 25 mM MgCl2 | 2.0 ul | HIV Probe5’FAM-CCACCCCACAAGATTTAAACACCATGCTAA-TAMRA 3′ |
| *Forward Primer (100 um) | 0.3 ul | |
| *Reverse Primer (100 um) | 0.3 ul | RCAS Forward Primer5′-GTCAATAGAGAGAGGGATGGACAAA-3′ |
| *Probe (100 um) | 0.05 ul | RCAS Reverse Primer5′-TCCACAAGTGTAGCAGAGCCC-3′ |
| TaqGold (5 U/ul) | 0.25 ul | RCAS Probe 5’FAM-TGGGTCGGGTGGTCGTGCC-TAMRA 3′ |
| Total | 20.0 ul |
Table 2. PCR master mix and primers for Single Copy Assay.
| cDNA cocktail/cDNA reactions | 1 RNA sample | First PCR cocktail | 1 plate (75 reactions) | Nested PCR cocktail | 1 plate (75 reactions) |
| 10X RT Buffer (Invitrogen) | 10 ul | 10X PCR buffer (Invitrogen) | 75 ul | 10X PCR buffer | 150 ul |
| 25 mM MgCl2 | 20 ul | 50 mM MgSO4 | 30 ul | 50 mM MgSO4 | 60 ul |
| 0.1M DTT | 1 ul | 10 mM dNTPs | 15 ul | 10 mM dNTPs | 30 ul |
| RNase-free water | 17.5 ul | 50 uM primers (ea) | 3 ul | 50 uM primers (ea) | 6 ul |
| Rnase-Out (Invitrogen) | 1 ul | Platinum Taq Hi Fi Enzyme (invitrogen) | 6 ul | Platinum Taq Hi Fi Enzyme (Invitrogen) | 12 ul |
| Superscript III (200 U/ul) (invitrogen) | 0.5 ul | Molecular-grade water | 468 ul | Molecular-grade water | 1086 ul |
Table 1. cDNA and PCR mixtures for Single Genome Sequencing.
| P6-RT 1 PCR program | Env 1 PCR program |
| 1. 94°C for 2 minute | 1. 94°C for 2 minute |
| 2. 94°C for 30 seconds | 2 .94°C for 30 seconds |
| 2 .94°C for 30 seconds | 3. 52°C for 30 seconds |
| 4. 72°C for 1 minute 30 seconds | 4. 72°C for 1 minute |
| 5. Go to #2, 44 cycles | 5. Go to #2, 44 cycles |
| 6. 72°C for 3 minutes | 6. 72°C for 3 minutes |
| 7. 4°C hold | 7. 4°C hold |
| P6-RT 2 PCR program | Env 2 PCR program |
| 1. 94°C for 2 minutes | 1. 94°C for 2 minutes |
| 2. 94°C for 30 seconds | 2. 94°C for 30 seconds |
| 3. 55°C for 30 seconds | 3. 56°C for 30 seconds |
| 4. 72°C for 1 minute | 4. 72°C for 45 seconds |
| 5. Go to # 1, 40 (41 cycles total) | 5. Go to # 1, 40 (41 cycles total) |
| 6. 72°C for 3 minutes | 6. 72°C for 3 minutes |
| 7. 4°C hold | 7. 4°C hold |
Table 4. Thermocycler conditions for the Single Genome Sequencing Assay.
| Reaction | Primer | Sequence |
| P6-RT cDNA | 3500- | 5′ CTATTAAGTATTTTGATGGGTCATAA 3′ |
| env cDNA | E115- | 5’AGAAAAATTCCCCTCCACAATTAA 3′ |
| P6-RT 1. PCR | 3500- | 5′ CTATTAAGTATTTTGATGGGTCATAA 3′ |
| P6-RT 1. PCR | 1849+ | 5′ GATGACAGCATGTCAGGGAG 3′ |
| P6-RT 2.PCR | 1870+ | 5′ GAGTTTTGGCTGAGGCAATGAG 3′ |
| P6-RT 2.PCR | 3410- | 5′ CAGTTAGTGGTATTACTTCTGTTAGTGCTT 3′ |
| env 1. PCR | E115- | 5’AGAAAAATTCCCCTCCACAATTAA 3′ |
| env 1. PCR | E20+ | 5’GGGCCACACATGCCTGTGTACCCACAG 3′ |
| env 2. PCR | E30+ | 5’GTGTACCCACAGACCCCAGCCCACAAG3′ |
| env 2. PCR | E125- | 5’CAATTTCTGGGTCCCCTCCTGAGG 3′ |
| P6-RT sequencing | 2030+ | 5’TGTTGGAAATGTGGAAAGGAAGGAC 3′ |
| P6-RT sequencing | 2600+ | 5’ATGGCCCAAAAGTTAAACAATGGC3′ |
| P6-RT sequencing | 2610- | 5’TTCTTCTGTCAATGGCCATTGTTTAAC3′ |
| P6-RT sequencing | 3330- | 5’TTGCCCAATTCAATTTTCCCACTAA 3′ |
| env sequencing | E30+ | 5’GTGTACCCACAGACCCCAGCCCACAAG3′ |
| env sequencing | E125- | 5’CAATTTCTGGGTCCCCTCCTGAGG 3′ |
Table 5. Primers for the Single Genome Sequencing Assay.
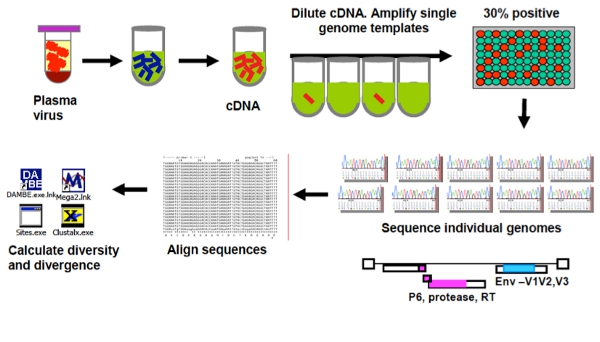
Figure 1. Overview of the Singe Genome Sequencing Assay (SGS).
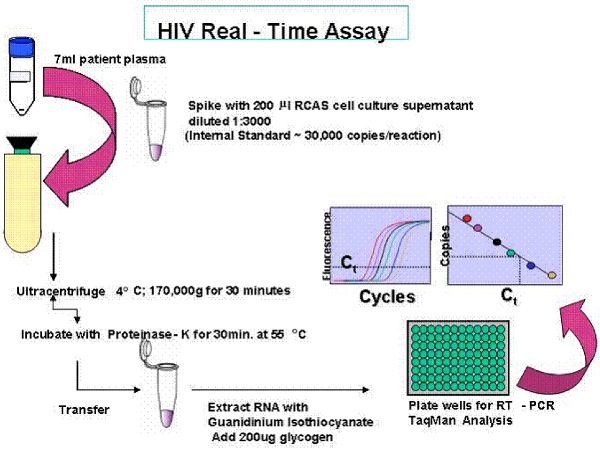
Figure 2. Overview of the Single Copy Assay (SCA).
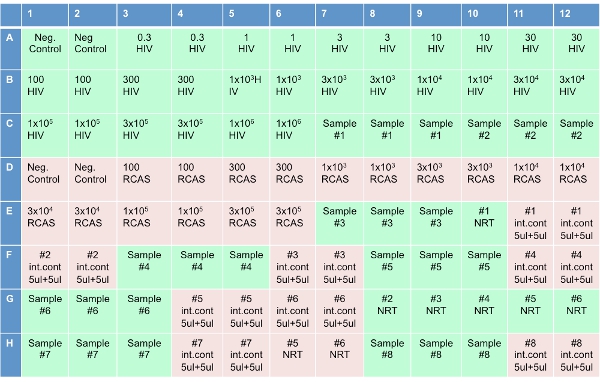
Figure 3. Plate set-up for the Single Copy Assay.
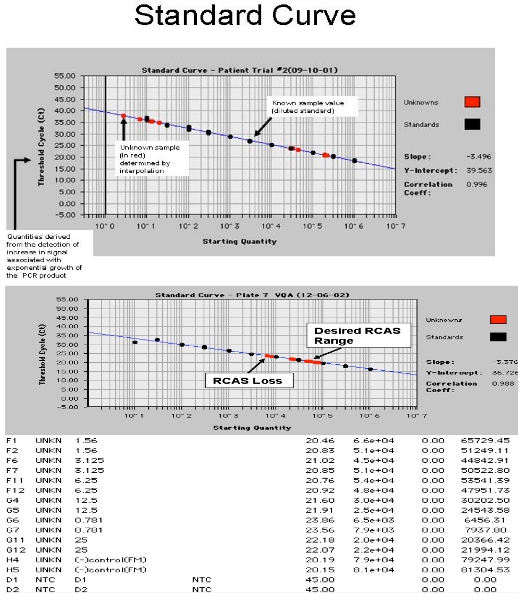
Figure 4. Screen shot from the ABI 7700. A. showing the HIV-1 RNA standard curve with patient samples and B. showing RCAS standard curve with spiked patient samples.
