Capabilities to develop and characterize conjugate nano-biological systems interfacing solid nanostructures and biological polymers are becoming increasingly important to further advances in next-generation bio-sensing and bio-actuation technologies1,2. This involves multi-disciplinary studies across a number of research fields, such as the fabrication of pertinent solid-state components (micro-or nano- electrodes, nano-engineered coatings, nanowires, or nanoparticles)2,3,4; immobilization of biomolecules on the surfaces to create desired bioconjugates5,6,7; and monitoring nano-biological interfaces1. In most cases, the selection of optimal fabrication, bio-functionalization, and characterization methods is strongly inter-related. Clearly, the choice of nanofabrication techniques would be driven by the requirements of the solid state components of the system, being largely dependent on the detection method, which in turn is determined by the nature of the biopolymers involved and the purpose of monitoring the interface.
Out of a broad variety of techniques applied to characterize bioconjugate systems1,3, surface enhanced Raman spectroscopy (SERS) has emerged as a highly promising method for the detection of chemical and biological species on surfaces8,9,10,11. SERS employs inelastic scattering of monochromatic light by surface-immobilized biomolecules (Figure 1) allowing the capture of unique signatures corresponding to molecular vibrations. This capability to distinguish among different molecules without involving labels, complex chemistry, or time-consuming steps, makes SERS a potentially very efficient method of bio-detection. Another important advantage of SERS is its high sensitivity. The excitation of localized surface plasmons by light interacting with noble metal nanostructures (SERS substrates) increases dramatically the intensity of Raman scattering by the analyte, allowing the detection of very small amounts of molecules, from monolayers down to the single-molecule limit8,9,10,11. Finally, most biomolecules require aqueous solutions to be stable. Because water often has limited Raman activity, background signal from aqueous samples is minimized9. Applications of SERS have exhibited an exponential increase over the last decade10. However, a much discussed challenge of SERS is that the electromagnetic enhancement of Raman scattering depends critically on the size, shape, and spacing of metal nanostructures where plasmonic waves are induced11,12,13. In order to achieve efficient and reproducible SERS measurements, control over the substrate geometry is required at the nanoscale dimensions.
Figure 1. Scheme of surface-enhanced Raman spectroscopy.
Numerous methods employed to fabricate SERS substrates11,12,13 can be roughly classified into bottom-up and top-down methods. Methods of the first type employ various processes of self-assembly or directed chemical synthesis to produce nanostructures. Often addressed examples include immobilization of monodisperse nanoparticles on solid supports11,12,13, thermal, sputter, or electrochemical deposition of roughened metal films11,12, and various chemical synthesis methods13. Although such techniques tend to be relatively simple and inexpensive, most of them are challenged by a lack of control over the location of the structures, and limited sample-to-sample reproducibility.
In contrast, top-down lithography techniques employ manipulable instruments such as particle beams to create desired patterns on surfaces. One of the most often used nanolithography methods, electron beam lithography (EBL), offers superb control over features down to below 10 nm and also a flexibility to allow for different substrate designs on solid supports11,12. In EBL, a beam of electrons focused down to a spot of a few nanometers in diameter scans across a surface of an electron sensitive material (resist) causing a chemical change in exposed regions. For positive tone resists such as polymethylmethacrylate (PMMA), electron beam exposure results in scission of the polymer chains composing the resist, leading to an increased solubility in an appropriate solvent (developer). The process of electron-beam lithography includes spin-coating of a uniform layer of resist on a substrate; exposure of the targeted resist material in a vacuum chamber with an electron beam; and development of the sample to remove the soluble regions.
Dielectric supports underneath metallic nanostructures, such as fused silica, have been shown to significantly increase the intensities in SERS due to localization of plasmonic waves compared to other materials such as silicon14,15. However EBL patterning on dielectric substrates, especially at the nanoscale, involves significant challenges due to charge build-up during exposure. Previously, we have shown16,17 that these difficulties can be overcome by placing conductive polymer layers above the resist. Figure 2 shows a schematic of the overall fabrication process using EBL exposure and development followed by metal deposition and liftoff to produce metallic nanostructures on fused silica supports. Please click here to view a larger version of this figure.

Figure 2. Scheme of electron beam lithography, metal deposition, and liftoff process steps employed to fabricate metallic nanostructures on dielectric substrates16-19. In this paper, we present the entire sequence of process steps involving SERS substrates fabrication by EBL, bio-functionalization of the substrates, and collection of the Raman spectra. Three designs explored in our recent works18,19, 20 are addressed (see Figures 3 and 4, and Table 1). In Design 1, recombinant protein A is immobilized on bio-functionalized Au nanostructures on a fused silica (FS) support18, and SERS detection of the protein is demonstrated. In Design 2, recombinant glucose-binding protein22,27,28 with and without the ligand (D-glucose) is immobilized by means of histidine tags in spaces between Ag nanostructures on Ni-coated FS, and the binding of glucose to the protein is detected. In Design 3, thiolated dopamine-binding DNA aptamer20,24 is immobilized on Au nanostructures on FS, and the binding of dopamine by immobilized aptamer is demonstrated. Inclusive of all relevant experimental steps from substrate preparation to Raman spectra acquisition, and representative of different biomolecules and strategies of immobilization, these examples are useful for a broad variety of applications, from exploration research interrogating nano-biological interfaces by SERS to the development of SERS biosensors of small molecules employing protein- or aptamer-ligand binding as a recognition method. Please click here to view a larger version of this figure.

Figure 3. Schemes of three representative designs using different biomolecules, methods of immobilization, and substrate materials: (A) protein A immobilized on the noble metal nano-dots functionalized by a self-assembled monolayer (SAM) of 11-mercaptodecanoic acid (MUA) in DI water; (B) histidine-tagged glucose binding protein (GBP) complexed with D-glucose immobilized on the substrate surface between noble metal nano-dots; (C) thiol-terminated dopamine binding aptamer completed with dopamine (DBA) immobilized on noble metal nano-dots. See further details in Table 1. In Design 2 illustrated by panel (B), a sample without the corresponding ligand was also prepared for comparison. Please click here to view a larger version of this figure.

Figure 4. Biomolecules employed in three designs: (A) protein A; (B) glucose binding protein and D-glucose; (C) dopamine binding DNA aptamer and dopamine. The protein tertiary structures in (a) and (b) are taken from Protein Data Bank, PDB ID 1BDD21 and 2HPH22, respectively, and drawn with VMD for LINUXAMD64, version 1.9.123. The aptamer secondary structure in (c) is predicted from the sequence24 using ValFold25 software and drawn with PseudoViewer 3.026. The letters G, A, T, and C correspond to guanine, adenine, thymine, and cytosine nucleotides, respectively. Please click here to view a larger version of this figure.
| Design 1 | Design 2 | Design 3 | |
| Biopolymer | Protein A | Glucose binding protein (GBP) | Dopamine binding aptamer (DBA) |
| Binder | 11-Mercaptoundecanoic acid (MUA) self-assembled monolayer (SAM) | Histidine tags | Thiol linkers |
| Ligand | None | D-glucose | Dopamine |
| Solution | Deionized (DI) water | Potassium phosphate buffer | Tris(hydroxymethyl)aminomethane (TRIS) and ethylenediaminetetraacetic acid (EDTA) buffer; Phosphate buffered saline (PBS) |
| Substrate | Au structures on FS | Ag structures on Ni-coated FS | Au structures on FS |
| Patterned area | 4 μm x 10 μm | 4 μm x 8 μm | 4 μm x 10 μm |
| Pattern | Au dots, 50 nm pitch | Ag dots, 40 nm pitch | Au hexagons, 200 nm pitch |
| Ag hexagons, 200 nm pitch | Au unstructured pads | ||
| Ag unstructured pads | |||
| EBL exposure doses | Dots: | Dots: 105 μC/cm2 | Hexagons: 180 μC/cm2 |
| Array I 120 μC/cm2 | Hexagons: 170 μC/cm2 | ||
| Array II 96 μC/cm2 | |||
| Array III 72 μC/cm2 | |||
| Laser excitation wavelength | 532 nm | 532 nm | 780 nm |
Table 1. Three designs of nano-biological systems.
Collecting control Raman spectra for the main components, including free proteins in solution and free ligands in solution or in powder form without using metal-containing substrates, is important to enable a proper comparison as well as for interpretation purposes. Figure 5A presents a typical Raman spectrum for free protein A in DI water on a glass slide without nanostructured substrates. Two bands with the highest Raman intensity, the band at 2,931 cm-1 and at 1,091 cm-1, correspond to vibrations involving C-H and C-S bonds, respectively. Other bands with a lower Raman intensity such as 563 cm-1, 1,450 cm-1, 1,653 cm-1 and 2,426 cm-1, can be attributed to a superposition of vibration modes18,33,34,35. Control Raman spectra for the ligand free GPB in buffer solution with three different concentrations, 0.3, 0.9 and 1.3 mM, are shown in Figure 5B. In the figure, the broad band around 3,400 cm-1 corresponds to the solvent36, whereas the band at 2,935 cm-1represents vibrations involving C-H bonds of the protein33,34. Figure 5C shows the high wavelength Raman spectrum for D-glucose in buffer solution for different concentrations: 1, 6, 100, 200, and 400 mM. When the concentration of glucose is increased, C-H bonds vibration bands arise at 2,890 cm-1 and 2,960 cm-1. Control Raman spectra of dopamine in crystal form obtained with both 532 nm and 780 nm excitation wavelengths are shown in Figure 5D. Much of the Raman spectrum comes from the benzene ring bending and C-H bond stretches of the molecule20,37. Some of the bands at around 3,000 cm-1 are only observed at the 532 nm but not 780 nm excitation wavelength.

Figure 5. Control Raman spectrum of protein A in DI water obtained at 532 nm excitation wavelength18 (A); Raman spectra of ligand-free glucose binding protein in buffer solution obtained at 532 nm excitation wavelength (B); Raman spectra of D-glucose in buffer solution obtained at 532 nm excitation wavelength (C); and spectra of dopamine powder obtained at 532 nm and 780 nm excitation wavelengths20(D). All the spectra are regular Raman spectra of solutions (a,b,c) and powder (d) on glass slides without nanostructured substrates. In (D), the assignment of Raman shift regimes to various molecular vibrations was done using General Atomic and Molecular Electronic Structure System (GAMESS)32 and MacMolPlt31 software as detailed elsewhere20. Reprinted panel (a) with permission from18 American Vacuum Society. Please click here to view a larger version of this figure.
In order to obtain SERS spectra for surface-immobilized biomolecules, substrates comprising metallic nanostructures on fused silica supports are fabricated as described in steps 1-3. The quality of fabricated substrates is monitored using scanning electron microscopy (SEM). The standard SEM procedures are described elsewhere16,17,18,19 and not included in the present protocol. Figure 6 shows representative SEM images of Au and Ag nano-dots and nano-hexagon like structures (panels a-d), as well as non-structured Au and Ag pads (panels e and f, respectively). The next steps involve immobilization of biological material on the nano-structured substrates employing three designs listed in Table 1, and acquisition of their SERS spectra. In order to maintain an aqueous environment during Raman imaging, each sample is placed in its corresponding solution (see Table 1), capped with a thin glass cover, and sealed as illustrated in Figure 7.

Figure 6. Scanning electron microscope (SEM) images of 10 nm thick Au and Ag surface nanostructures employed as SERS substrates: (A,B) arrays of nanodots; (C,D) arrays of nano-hexagons; (E,F) unstructured pads. Substrates with Au structures (left) employ FS supports, whereas substrates with Ag structures (right) use a 10 nm thick Ni coating on the FS. The images were obtained as described elsewhere16,17,18. Please click here to view a larger version of this figure.

Figure 7. Scheme of water-proof chamber for Raman imaging of bio-functionalized samples in solution.
In Design 1, recombinant protein A is immobilized on the substrates functionalized by a self-assembled monolayer of 11-mercaptodecanoic acid (MUA) in DI water18. The substrates in this design comprise three arrays of Au dots with a 50 nm pitch and varying inter-dot distances on fused silica (see Figures 6A and 8). The process of protein immobilization starts with the formation of a SAM on the substrates. To obtain covalent binding between the SAM and the protein, the carboxylic acid groups of SAMs are transformed into amine reactive NHS-ester by treatment with a mixture of N-ethyl-N'-(3-(dimethylamino) propyl) carbodiimide (EDC) solution and N-hydroxysuccinimide (NHS) solution in DI water. Immobilization of protein A occurs by displacement of the NHS group by lysine residues of the protein38. An example of imaging samples for Design 1 with protein A immobilized on Au nanostructures is shown in Figure 9. Figure 9A presents an optical microscope image of the samples, which comprise three arrays of bio-functionalized Au nanodots with different inter-dot gaps (see also Figures 3Aa and 8), and Figure 9B shows the Raman spectral mapping over these arrays. It can be seen that the highest Raman intensities are found for Array I where the inter-dot gaps are the narrowest, whereas lower intensities are obtained for Array III with the widest inter-dot gaps. This can be explained by a stronger plasmon coupling effect produced by higher electric fields in the narrow spaces between the dots18. Figure 9C shows the strongest SERS spectra obtained for Arrays I and II. The spectra exhibit several bands (1,630 cm-1, 1,964 cm-1, 2,280 cm-1, 2,577 cm-1, and 2,916 cm-1) in proximity to the Raman modes of free protein A in solution seen in Figure 5A. Attributable to vibrations of various bonds found in proteins, these bands either appear at similar locations in both immobilized protein and in solution, or are slightly shifted to somewhat higher wavenumbers when immobilized. In contrast, SERS spectra of similar nanostructured substrates functionalized by MUA SAM without the protein show an entirely different pattern18, confirming that Figure 9 represents SERS mapping of surface-immobilized protein A. Please click here to view a larger version of this figure.

Figure 8. SEM images of three arrays of Au nanodots on FS substrate used in Design 118. The arrays have the same 50 nm pitch and slightly different dot radii resulting in different widths of inter-dot gaps. This is achieved by applying different EBL exposure doses to produce PMMA masks for the three arrays (see Table 1). Higher exposure doses result in wider holes in PMMA masks, allowing for larger Au dot sizes after metallization and liftoff. Reprinted with permission from18 American Vacuum Society. Please click here to view a larger version of this figure.

Figure 9. SERS imaging of substrate-immobilized protein A in Design 118. (A) Optical microscope image of the sample, comprising three arrays of Au nanodots with 50 nm pitch and different inter-dot gaps on a fused silica substrate (see also Figure 6), bio-functionalized as shown in Figure 3A. (B) Raman mapping of the sample. (C) SERS spectra from dot Array I and II. In panel (B), the vertical axis represents the distance across the substrate, the horizontal axis represents the Raman shift, and the legend bar indicates the Raman intensities. Vertical dashed lines in panels (B) and (C) represent benchmark Raman bands of free protein A in solution and (*) in panel (C) indicates SERS bands from Array I. The Raman spectra were obtained 532 nm excitation wavelength. Reprinted with permission from18 American Vacuum Society. Please click here to view a larger version of this figure.
In Design 2, recombinant glucose binding protein (GBP)22 complexed with D-glucose (ligand) is immobilized on the appropriate substrates in potassium phosphate buffer solution. Samples with immobilized ligand-free GBP are also prepared for comparison. In this design, glucose-binding protein is attached to surface by means of a histidine tag, which binds well to Ni but not to noble metals6. Since the substrates comprise arrays of Ag nano-dots, nano-hexagons, and unstructured Ag pads on Ni-coated FS (Figures 6B, 6D, and 6F, respectively), one can expect most of immobilized protein molecules to be located in gaps between Ag nanostructures where Ni coating is available. The Raman spectra obtained for immobilized glucose-free and glucose-bound GBP are shown in Figures 10A and 10B, respectively. All these spectra exhibit a broad band at approximately 3,300 cm-1, which corresponds to the buffer solution36. The spectra obtained with an unstructured Ag pad contain only this single band and do not show any protein vibration modes, confirming that immobilized protein is not found on the Ag surface as expected. In contrast, the spectra obtained with arrays of Ag nano-dots and nano-hexagons exhibit bands around 1,550 cm-1 and 2,900 cm-1, which represent the analyte33,34. In particular, the broad band around 1,550 cm-1, known as the amide II band, is attributable to peptide bonds vibrations in proteins34,35. In the case considered, this band represents a superposition of the vibration modes from GBP immobilized on Ni surface between Ag features, and is indicative of SERS enhancement of these modes in the vicinity of noble metal nanostructures when the substrates containing nano-dots or nano-hexagons are used. This band is very weak for the protein in solution in the absence of SERS enhancement (Figure 5B) and absent on Ag pads without Ni surface available for the protein binding, but it is well pronounced for nanostructured substrates with some Ni surface accessible for the protein to bind. However, even more important for the present study are the other, narrower bands around approximately 2,900 cm-1 that can be attributed to C-H bond wibrations33,34. The spectrum of glucose-free GBP shows a pronounced band at 2,933 cm-1 with the nano-dots substrate, and a weak but discernible band at a similar wavelength with the nano-hexagons substrate (Figure 10A). Distinct from the case of glucose free protein, the SERS spectra of glucose-bound GBP shown in Figure 10B exhibit two bands corresponding to C-H bonds vibrations regimes, at 2,850 cm-1 and 2,910 cm-1. The bands are well pronounced in the spectrum of glucose-bound GBP on nano-hexagons substrate, and they also can be seen in the spectrum of GBP on nano-dots substrate. The band at 2,850 cm-1 is reasonably close to the 2,890 cm-1 one in the control Raman spectrum from D-glucose in solution, and therefore it can be attributed to glucose bound to the protein, whereas the other band (at 2,910 cm-1) is attributable to C-H bond vibrations of both the protein and glucose. One can conclude that difference of SERS signatures from glucose-free and glucose-bound substrate-immobilized GBP is observable in this region, and C-H bond vibrations of protein-bound glucose are detectable employing the design described.
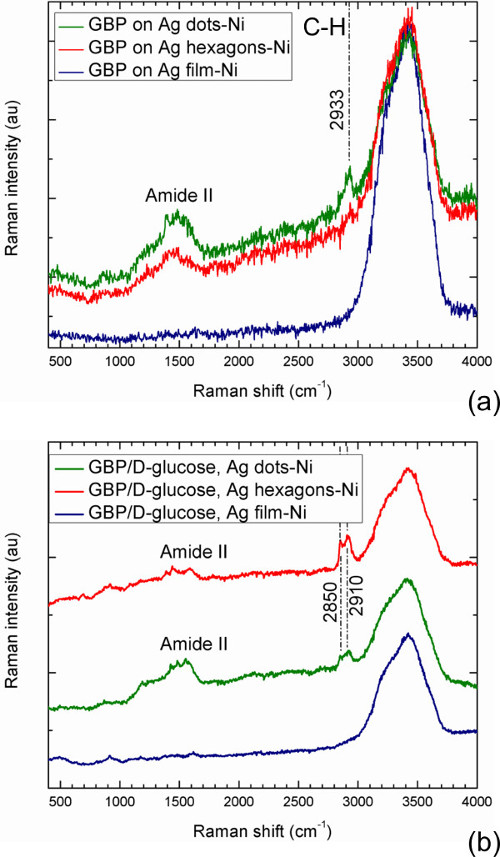
Figure 10. SERS spectra of ligand-free (A) and ligand-bound (B) glucose-binding protein immobilized on three different substrates in Design 2. The spectra were obtained with 532 nm excitation wavelength. Please click here to view a larger version of this figure.
In Design 3, the customized dopamine binding aptamer (DBA) with thiol termination24 is immobilized on the substrate in tris(hydroxymethyl)aminomethane (TRIS) ethylenediaminetetraacetic acid (EDTA) buffer solution, and the dopamine is then bound to the immobilized aptamer20. The substrates for this design contain arrays of Au nano-hexagons on FS (Figure 6C). Unstructured Au pads (Figure 6E) are also used for control purposes. Since DNA is intrinsically fluorescent39, 780 nm excitation wavelength is used in Design 3 to reduce this factor. In this design, the recognition element (aptamer) is not Raman active in the region of Raman shifts considered in Figure 11, whereas dopamine shows a significant Raman activity in this region. Since signal from samples exposed to only dopamine without immobilized aptamer shows no resultant dopamine bands20, the observed SERS bands are expected to originate from aptamer-bound dopamine. Figure 11 compares the SERS spectra of immobilized aptamer on gold nanostructures before and after the addition of dopamine. As expected, immobilized dopamine-free aptamer does not exhibit Raman bands. In contrast, a number of pronounced Raman bands are observed for dopamine-bound immobilized aptamer. The positions of most bands in Figure 11 are close to those in crystalline dopamine, albeit with differences in amplitudes.

Figure 11. SERS spectra of ligand-free (purple line) and ligand-bound (blue line) dopamine-binding aptamer immobilized on Au nano-hexagon substrates in Design 320. The red line shows a control SERS spectrum of dopamine powder. Please click here to view a larger version of this figure.
