The RAF family kinases have both catalytic and allosteric activities, which enable their disease-related mutants to turn on the downstream signaling through different mechanisms13,14,16,17,18. The constitutively active RAF mutants directly phosphorylate their substrates, while the kinase-dead RAF mutants fulfill their function through transactivating wild-type counterparts. As shown in Figure 1B, both constitutively active RAF mutants (such as Regulatory spine (R-spine) mutants (BRAF(L505M), CRAF(DDEE/L397M), and ARAF(DGEE/L358M))13,23,25,26, BRAF(V600E), and BRAF(∆NVTAP)) and kinase-dead RAF mutants (such as Catalytic spine (C-spine)-fused BRAF(∆NVTAP/V471F)15) activates ERK when expressed in 293T cells. Therefore, the ability of RAF mutants to activate ERK signaling in cells cannot serve as a standard to distinguish a constitutively active mutant from a kinase-dead mutant, although some kinase-dead mutants with moderate dimer affinity/stability turns on downstream signaling only with the cooperation of active Ras. Three methods that we presented here can help us effectively characterize all disease-related RAF mutants. The biological properties of all RAF mutants revealed by using these assays in our previous studies have been summarized in Table 1.
The first method that we can use to distinguish the constitutively active RAF mutants from the kinase-dead RAF mutants is the in vitro kinase assay. In this assay, the RAF mutants were purified by immunoprecipitation and the catalytic reactions were carried out in vitro with kinase-dead MEK1(K97A) and ATP as substrates. The catalytic activity of RAF mutants was measured as the AL(Activation Loop)-phosphorylation of MEK1(K97A) in the kinase reaction mixtures by immunoblot. As shown in Figure 1C, this assay can effectively probe the catalytic activity of R-spine mutants of BRAF, CRAF and ARAF13,15,23, BRAF(V600E)9, and BRAF(∆NVTAP)15, but not that of kinase-dead BRAF(V471F/∆NVTAP)15 that has a fused C-spine. However, the catalytic activity of constitutively active RAF mutants with weak dimer affinity/stability such as ARAF R-spine mutant (ARAF(DGEE/L358M)) (Figure 1C, lane 4), CRAF and ARAF mutants with altered dimer interface (CRAF(DDEE/L397M/R401H), ARAF(DGEE/L358M/APE/R362H))15,22 (Figure 1D,E) might not be probed by using this assay, since their dimers were broken during the purification, especially when the purification was carried out with buffers containing strong or high-concentration detergents. To avoid the loss of catalytic activity of RAF mutants with weak dimer affinity/stability, we usually fused these mutants with GST (glutathione S–transferase), a dimeric protein with strong affinity/stability before carrying out in vitro kinase assay, which can rescue the catalytic activity of these RAF mutants15,22 (Figure 1E). In general, most RAF mutants could be classified as constitutively active or kinase-dead mutants by using this assay.
The second method that we described here is the RAF co-activation assay that can be used to evaluate the allosteric activity of kinase-dead RAF mutants13,15,21,22,23,25. Although a few kinase-dead RAF mutants with very high dimer affinity/stability such as BRAF(V471F/∆NVTAP)15, can directly activate endogenous RAF molecules when expressed in cells (Figure 1B, lane 7), most kinase-dead RAF mutants require the cooperation of active Ras to transactivate wild-type RAFs. However, active Ras is a direct activator of ERK signaling, whose introduction will increase the basal level of active ERK. To avoid the interference of active Ras, we used N-terminus-truncated RAF mutants in this assay (Figure 2A). As shown in Figure 2B, kinase-dead mutants of BRAF, CRAF, and RAF with acidic NtA motif and C-spine fusion (BRAF(V471F, aa431-766), CRAF(DDEE/V363F, aa323-648), and ARAF(DGEE/V324F, aa284-606)) functioned as allosteric activators to trigger the catalytic activity of CRAF with non-phosphorylatable NtA motif (CRAF receiver, CRAF(AAFF, aa323-648)) when co-expressed in 293T cells. In contrast, the kinase-dead BRAF mutant (V471F/∆NVTAP, aa431-766) that have very high dimer affinity/stability (see below) turned on ERK signaling by triggering endogenous RAFs, even without the co-expression of CRAF receiver. Overall, this assay can be used to evaluate the transactivation ability of all kinase-dead RAF mutants.
The dimerization of RAF family kinases not only regulates their ability to activate downstream MEK-ERK signaling but also determines their sensitivity to RAF inhibitors15,16,20,22,24,27,28,29,30,31,32. In contrast to other protein-protein interactions among this pathway33,34,35, the dimerization of RAF family kinases and their mutants is relatively weak and difficult to be evaluated by using traditional biochemistry assays such as co-immunoprecipitation. To resolve this problem, we recently developed a complimentary split luciferase assay for measuring the relative dimer affinity/stability of RAF family kinases and their mutants15,22,36. As shown in Figure 3A, the RAF kinase domains (aa431-766 for BRAF, aa323-648 for CRAF, and aa284-606 for ARAF) were fused respectively with N-terminus(aa2-416) or C-terminus(aa398-550) of firefly luciferase (Nluc and Cluc), which will be brought together to assemble intact luciferase upon RAF dimerization. The activity of luciferase in this assay directly correlates with the quantity of RAF dimers. This assay is very sensitive and can detect even a very weak dimerization of RAF mutants. As we reported before15, the oncogenic BRAF(V600E) functions as a dimer, and only a compound mutation with the Arg-to-His mutation(R509H) in dimer interface and the non-canonic APE alteration(P622A in APE motif) can completely abolish its dimerization and thereby block its catalytic activity (Figure 3B). By using in vitro kinase assay, we further found that BRAF(V600E) variant with the altered APE motif, BRAF(V600E/AAE), but not that with the Arg-to-His mutation in dimer interface, BRAF(V600E/R509H), lost its catalytic activity upon purification (Figure 3C), suggesting that the former variant has much less dimer affinity/stability than the latter. To directly measure the propensity of these BRAF(V600E) variants to form dimers, we carried out a complimentary split luciferase assay, and confirmed that these variants had quite different dimer affinity/stability with BRAF(V600E) higher than BRAF(V600E/R509H) than BRAF(V600E/AAE) than BRAF(V600E/R509H/AAE) (Figure 3D). The luciferase signal produced by monomeric BRAF(V600E/R509H/AAE) was comparable with that of non-transfected 293T control (Figure 3D, left panel lane 5), indicating that this assay has a very low background. To further determine the effectiveness of complimentary split luciferase assay, we next measured by using this assay the relative dimer affinity/stability of a group of oncogenic BRAF mutants with in-frame deletions of β3- αC loop that was identified by us and other groups15,37,38. As we have known, although all of these mutants activated ERK signaling when expressed in 293T cells (Figure 3E), they exhibited quite differential catalytic activity in vitro (Figure 3F). The BRAF(∆MLN(aa484-486 del)) and BRAF(∆NVTAP(aa486-490 del)) were very active upon purification by immunoprecipitaion, whereas the BRAF(∆NVTAPT(aa486-491 del)) and BRAF(∆QA(aa496-497 del)) lost their catalytic activity under same condition though it could be rescued by GST fusion. In addition, the kinase-dead version of BRAF(∆NVTAP), BRAF(V471F/∆NVTAP) exhibited a strong potency to activate endogenous RAFs when expressed in cells (Figure 1B, lane 7). These data suggest that this group of BRAF mutants have quite different dimer affinity/stability with BRAF(∆NVTAP) higher than BRAF(∆MLN) than BRAF(∆NVTAPT) and BRAF(∆QA). Indeed, the result from the complimentary split luciferase assay completely supported our inference (Figure 3G). Taken together, our data indicate that the complimentary split luciferase assay is a reliable method to measure the relative dimer affinity/stability of RAF mutants.
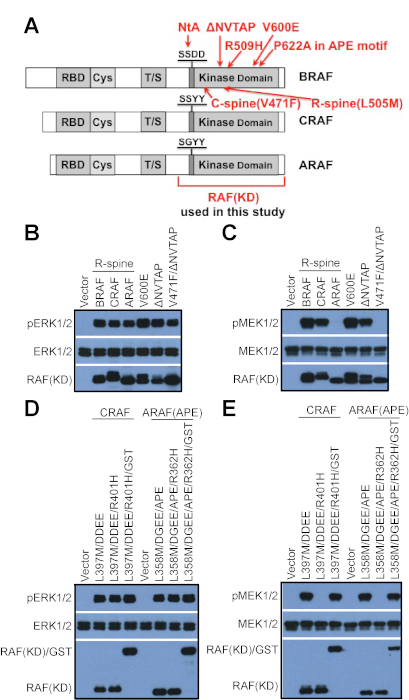
Figure 1: Probe the catalytic activity of RAF mutants by using in vitro kinase assay. (A) A schematic diagram for RAF mutations used in this study. (B) Both constitutively active and kinase-dead RAF mutants can activate ERK signaling when expressed in cells. 293T cells were transfected with vectors that encode different RAF mutants, and the activity of ERK was detected by anti-phospho-ERK1/2 immunoblot. (C) Constitutively active RAF mutants with low dimer affinity/stability as well as kinase-dead RAF mutants cannot phosphorylate MEK in vitro upon purification by immunoprecipitation. RAF mutants in B were purified by using anti-FLAG beads, and their catalytic activity was probed by using in vitro kinase assay as described in the protocol. (D-E) The in vitro catalytic activity of constitutively active RAF mutants with low dimer affinity/stability can be rescued by GST fusion. (D) Constitutively active RAF mutants and their GST-fused counterparts were expressed in 293T cells and their activity was measured by anti-phospho-ERK1/2 immunoblot. (E) RAF mutants in D were purified by immunoprecipitation, and their in vitro catalytic activity was probed by using in vitro kinase assay as described in the protocol. RAF R-spine mutants: BRAF(L505M), CRAF(DDEE/L397M), and ARAF(DGEE/L358M). "∆NVTAP" represents for the deletion of aa486-490 in BRAF. The APE mutation of ARAF means A475P that generates a canonical APE motif in ARAF. "KD" represents for "kinase domain" in the full text. BRAF(KD) means the aa431-766 fragment of BRAF, while CRAF(KD) and ARAF(KD) represent respectively for the aa323-648 fragment of CRAF and the aa284-606 fragment of ARAF. The results in this figure have been reported previously15,22. Please click here to view a larger version of this figure.
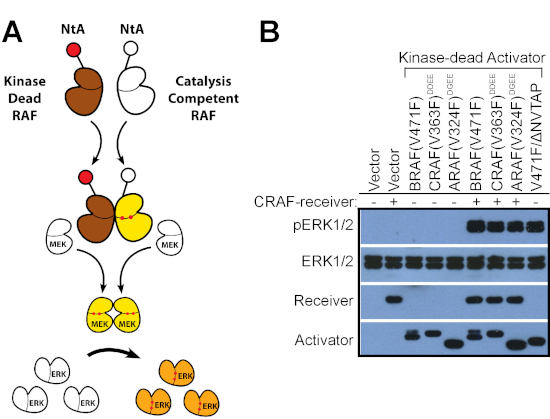
Figure 2: Evaluate the ability of RAF mutants to transactivate wild-type RAFs by using the RAF co-activation assay. (A) A diagram illustrating the RAF co-activation assay. (B) Kinase-dead RAF mutants with acidic NtA motif were co-expressed with catalysis-competent CRAF receiver in 293T cells, and the activity of ERK1/2 in 293T transfectants was measured by anti-phosho-ERK1/2 immunoblot. Most kinase-dead RAF mutants except BRAF(V471F/∆NVTAP) that has a very high dimer affinity/stability required the co-expression of exogenous RAF receiver to turn on ERK signaling. The results in this figure have been reported previously15,22. Please click here to view a larger version of this figure.
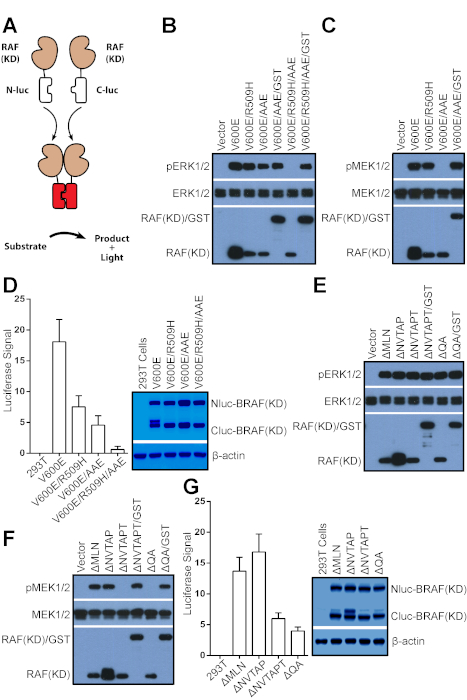
Figure 3: Measure the relative dimer affinity/stability of RAF mutants by using the complimentary split luciferase assay. (A) A diagram illustrating the complimentary split luciferase assay. (B-D) The dimerization of BRAF(V600E) variants is required for their catalytic activity in vivo and in vitro. (B) The monomeric BRAF(V600E) variant, BRAF(V600E/R509H/AAE) has no catalytic activity in vivo, which can be recovered by GST fusion. BRAF(V600E) variants and their GST-fused counterparts were expressed in 293T cells, and their activity was measured by anti-phospho-ERK1/2. (C) The BRAF(V600E) variant with low dimer affinity/stability, BRAF(V600E/AAE) lost its catalytic activity in vitro upon purification, which can be rescued by GST fusion. BRAF(V600E) variants from B were purified by immunoprecipitation and their catalytic activity was probed by in vitro kinase assay as described in the protocol. (D) The relative dimer affinity/stability of BRAF(V600E) variants was measured by using the complimentary split luciferase assay as described in the protocol. (E-G) BRAF mutants with in-frame deletion of β3- αC loop function as dimers to activate ERK signaling. (E) BRAF mutants with in-frame deletion of β3- αC loop activate ERK signaling when expressed in cells. BRAF mutants were expressed in 293T cells and their activity was measured as described in B. (F) BRAF mutants with low dimer affinity/stability from E lost their catalytic activity in vitro, which can be rescued by GST fusion. The in vitro catalytic activity of BRAF mutants and their GST-fused counterparts from E was measured as in C. (G) BRAF mutants with in-frame deletion of β3- αC loop have quite different dimer affinity/stability. The relative dimer affinity/stability of BRAF mutants with in-frame deletion of β3- αC loop was measured as in D. Error bars in D&G represent s.d. to show variance between independent experiments (n = 4). The AAE mutation of BRAF means P622A in APE motif that generates a non-canonical APE motif. "∆MLN", "∆NVTAPT", and "∆QA" represent respectively for the deletions of aa484-486, aa486-491, aa496-497 in the β3- αC loop of BRAF. Some results in this figure have been reported previously15. Please click here to view a larger version of this figure.
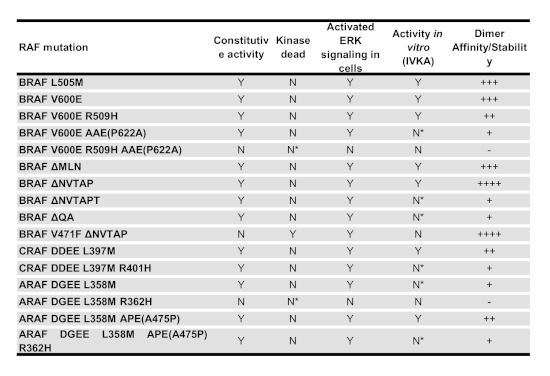
Table 1: The biological property of various RAF mutants. The catalytic activity, allosteric activity and relative dimer affinity/stability of all RAF mutants used in this study have been summarized in this table. "Y" means "Yes", while "N" stands for "No". "N*" indicates that those mutants have catalytic activity if fused with GST. "++++", "+++", "++", "+", and "-" represent respectively for "very strong", "strong", "intermediate", "weak", and "none".
