Studying site-directed DSB-induced repair processes in cells can be achieved via either stable or transient transfection. However, it should be noted that stable transfection ensures a homogenous cell population, which gives a unified and thus more reliable cellular response. In the case of transient transfection, only a small proportion of the cell population takes up and maintains the plasmid, which introduces diversity into the experiment. Establishing ER-I-PpoI or ER-AsiSI endonuclease-based cell systems require a 50% confluent cell population, which is more effectively transfected with plasmids encoding the endonuclease. For transfection, commercially available transfection reagents or viral infection-based methods can also be used. If a microscopic visualization technique is to be applied and transient transfection is required, the directed DSBs can be induced by 4-OHT addition 24 h after transfection, which binds to the ER-fused endonucleases and allows the nuclear translocation and DSB induction. To determine the most appropriate time-points, immunofluorescence-based microscopy and western blot detection of γH2AX at different time-points following 4-OHT treatment can be performed. Under physiological conditions, a maximum of 10-15 γH2AX foci per cell can be detected, and the formation of strong repair foci can be triggered by endonucleases (or various other techniques not discussed here e.g., laser microirradiation). A typical I-PpoI endonuclease leads to the formation of elevated γH2AX signals around the nucleolus by inducing DSBs at ribosomal DNA (rDNA). If the breaks are repaired by NHEJ or HR, the number of repair foci decreases over time. For this reason, representative time-points at 0 h, 30 min, 1, 2, 4, and 8 h following 4-OHT treatments are recommended. To track DNA repair processes, the most commonly used cell line is U2OS, as all known repair pathways are fully functional in these cells. When investigating several proteins in the same cells, co-localization can be studied by combining antibodies conjugated with different fluorophores with different emission wavelengths raised in different animal species as shown in Figure 1. Therein, the induction of DSBs via an inducible stable cell line is represented which is based on the ER-AsiSI restriction endonuclease fused with hemagglutinin tag (HA). Doxycycline can induce the expression and sequestration of HA-ER-AsiSI in the cytoplasm which can be tracked using an antibody against HA (Figure 1. third column, second raw). Incubation for 4 h with 4-OHT, 24 h after doxycycline addition, can induce high number of DSBs since the endonuclease has been translocated to the nucleus (Figure 1. third column, third raw and second column, third raw). DSBs can be visualized by using an antibody recognizing γH2AX.
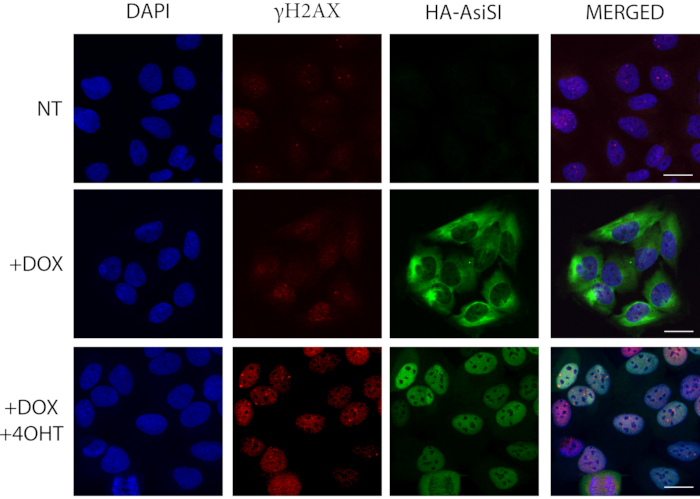
Figure 1: Immunofluorescence microscopy is used to detect γH2AX in cultured cells expressing HA-ER-AsiSI. DOX (doxycycline) addition activates the cytoplasmic expression of HA-ER-AsiSI and 4-OHT (4-hydroxytamoxifen) (4 h) induces the nuclear translocation of the fusion protein, which leads to the induction of DSBs at known genomic positions. HA (hemagglutinin) staining (anti-HA antibody) represents the HA-ER-AsiSI fusion protein in green, and the induction of breaks is verified by γH2AX staining (anti-γH2AX antibody) in red. Scale bars represent 20 µm. Please click here to view a larger version of this figure.
Co-localization of repair proteins at the damage site indicates that they are recruited to the same DNA lesion site, but they do not necessarily interact with each other. The resolution of the confocal microscopy is approximately 300 nm; to determine the binding pattern of specific repair proteins at the break site, super-resolution microscopy (STORM) is instead recommended 24. However, this method requires expensive microscopic equipment and an expert researcher. Alternatively, the binding pattern of the repair proteins can be examined by chromatin immunoprecipitation using DIvA or U2OS-pEP15 stable cell lines, which can express AsiSI and I-PpoI endonucleases in a regulated fashion, respectively17,21. Upon 4-OHT addition, both endonucleases can cut the DNA in a sequence specific manner which provides us the opportunity to design locus specific primers to the expected break sites and their surrounding genomic regions. By applying γH2AX antibody in the immunoprecipitation part of the ChIP, we can temporally follow the DNA repair kinetics upon different conditions (such as silencing or inhibition of certain repair factors of interest, i.e., DNA-PKcs). A typical experimental result obtained using ChIP-qPCR is represented in Figure 2. Therein, the temporal enrichment of γH2AX is demonstrated as a response to I-PpoI-induced DNA damage. On the left part of the image, the timely detected γH2AX signal is shown at the break site while on the right part, the γH2AX distribution is represented at a control gene region at which DSBs have not been induced.
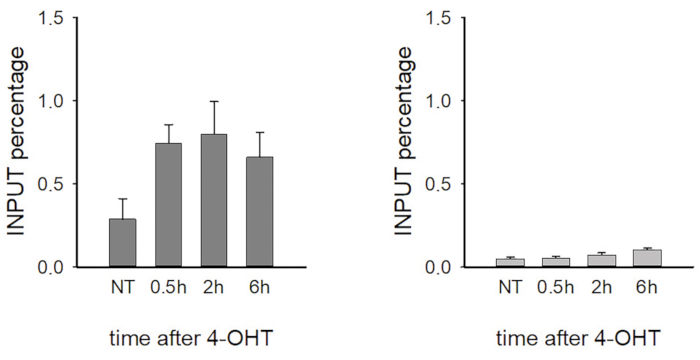
Figure 2: Temporal enrichment of γH2AX as determined by chromatin immunoprecipitation in response to I-PpoI-induced DNA damage. Left, γH2AX signal at the break site; right, γH2AX distribution at a control region where DSBs were not induced (anti-γH2AX antibody). The represented results are derived from one biological experiment, and error bars indicate the variations of the corresponding sample replicates. N=3. Please click here to view a larger version of this figure.
