Guide d’examen de la formation de graisse intramusculaire et de son origine cellulaire dans le muscle squelettique
Summary
Le remplacement du tissu musculaire sain par de la graisse intramusculaire est une caractéristique importante des maladies et affections humaines. Ce protocole décrit comment visualiser, imager et quantifier la graisse intramusculaire, permettant l’étude rigoureuse des mécanismes sous-jacents à la formation de graisse intramusculaire.
Abstract
Les progéniteurs fibro-adipogènes (PAF) sont des cellules stromales mésenchymateuses qui jouent un rôle crucial lors de l’homéostasie et de la régénération des muscles squelettiques. Les PAF construisent et maintiennent la matrice extracellulaire qui agit comme un échafaudage de myofibre moléculaire. De plus, les PAF sont indispensables à la régénération des myofibres car ils sécrètent une multitude de facteurs bénéfiques détectés par les cellules souches musculaires (MuSC). Dans les états malades, cependant, les FAP sont l’origine cellulaire de la graisse intramusculaire et du tissu cicatriciel fibrotique. Cette fibrose graisseuse est une caractéristique de la sarcopénie et des maladies neuromusculaires, telles que la dystrophie musculaire de Duchenne. Une barrière importante pour déterminer pourquoi et comment les PAF se différencient en graisse intramusculaire est la préservation efficace et la visualisation ultérieure des adipocytes, en particulier dans les sections de tissus congelés. Les méthodes conventionnelles de traitement des tissus musculaires squelettiques, telles que la congélation instantanée, ne préservent pas correctement la morphologie des adipocytes individuels, empêchant ainsi une visualisation et une quantification précises. Pour surmonter cet obstacle, un protocole rigoureux a été développé qui préserve la morphologie des adipocytes dans les sections musculaires squelettiques permettant la visualisation, l’imagerie et la quantification de la graisse intramusculaire. Le protocole décrit également comment traiter une partie du tissu musculaire pour la RT-qPCR, permettant aux utilisateurs de confirmer les changements observés dans la formation de graisse en observant les différences dans l’expression des gènes adipogènes. De plus, il peut être adapté pour visualiser les adipocytes par immunofluorescence de montage entier d’échantillons musculaires. Enfin, ce protocole décrit comment effectuer un traçage génétique de la lignée des FAP exprimant Pdgfrα pour étudier la conversion adipogénique des FAP. Ce protocole produit systématiquement des images immunofluorescentes à haute résolution et morphologiquement précises des adipocytes, ainsi qu’une confirmation par RT-qPCR, permettant une visualisation et une quantification robustes, rigoureuses et reproductibles de la graisse intramusculaire. Ensemble, le pipeline d’analyse décrit ici est la première étape pour améliorer notre compréhension de la façon dont les PAF se différencient en graisse intramusculaire et fournit un cadre pour valider de nouvelles interventions visant à prévenir la formation de graisse.
Introduction
L’infiltration de tissus musculaires sains avec fibrose graisseuse est une caractéristique importante de la dystrophie musculaire de Duchenne (DMD) et d’autres maladies neuromusculaires, ainsi que de la sarcopénie, de l’obésité et du diabète 1,2,3,4,5,6,7,8,9,10 . Bien que l’augmentation de l’infiltration de graisse dans ces conditions soit fortement associée à une diminution de la fonction musculaire, notre connaissance du pourquoi et de la façon dont la graisse intramusculaire se forme est encore limitée. Les FAP sont une population de cellules stromales mésenchymateuses multipotentes présentes dans la plupart des organes adultes, y compris le muscle squelettique11,12. Avec l’âge et dans les maladies chroniques, cependant, les FAP produisent du tissu cicatriciel fibrotique et se différencient en adipocytes, qui sont situés entre les myofibres individuels et forment de la graisse intramusculaire 13,14,15,16,17,18,19,20.
Pour commencer à lutter contre la formation de graisse intramusculaire, les mécanismes de transformation des PAF en adipocytes doivent être définis. PDGFRα est le marqueur « de référence » dans le domaine pour identifier les FAP dans le muscle de plusieurs espèces 13,16,17,18,20,21,22,23,24,25,26,27. En conséquence, plusieurs lignées Cre murines inductibles au tamoxifène, sous le contrôle du promoteur Pdgfrα, ont été générées, permettant de manipuler génétiquement les PAF in vivo à l’aide du système Cre-LoxP 27,28,29. Par exemple, en combinant cette lignée Cre inductible avec un rapporteur génétique, le traçage de la lignée des PAF peut être effectué, une stratégie que nous avons appliquée avec succès à la carte du destin des PAP dans le tissu adipeux musculaire et blanc20,30. Outre le traçage de la lignée, ces lignes Cre fournissent des outils précieux pour étudier la conversion FAP en graisse.
Un obstacle majeur dans la définition du mécanisme de conversion adipogénique des FAP en graisse intramusculaire est la capacité de quantifier rigoureusement et de manière reproductible la quantité de graisse intramusculaire qui s’est formée dans différentes conditions. La clé est d’équilibrer la préservation des tissus musculaires et adipeux et de l’associer aux méthodes de coloration disponibles pour visualiser les adipocytes. Par exemple, le muscle squelettique est souvent congelé sans fixation préalable, préservant les myofibres mais perturbant la morphologie des adipocytes (Figure 1). En revanche, la fixation suivie de l’incorporation de paraffine, tout en affichant la meilleure histologie tissulaire, y compris les adipocytes, élimine tous les lipides, rendant ainsi la plupart des colorants lipophiles, tels que le colorant couramment utilisé Oil Red O, inutilisables.
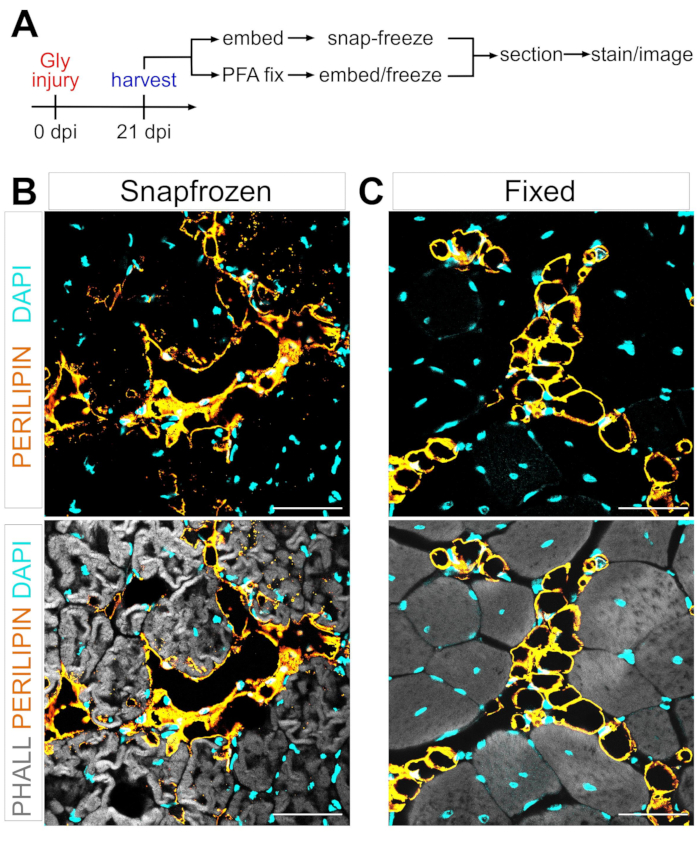
Figure 1 : Images représentatives de la graisse intramusculaire dans les tissus musculaires congelés par rapport aux tissus musculaires fixes. (A) Aperçu schématique de la configuration expérimentale. Images immunofluorescentes montrant des adipocytes (jaunes), des myofibres (gris) et des noyaux (cyan) dans les AT (B) congelés et (C) fixes 21 jours après une lésion au glycérol. Barres d’échelle : 50 μm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
Le protocole décrit ici préserve la morphologie des myofibres et des adipocytaires et permet la visualisation et l’analyse de plusieurs types de cellules. Cette approche est basée sur la coloration par immunofluorescence des adipocytes dans le tissu musculaire fixé au paraformaldéhyde (PFA), ce qui permet une co-coloration avec plusieurs anticorps. Il peut également être facilement adapté pour afficher spatialement la graisse intramusculaire dans les tissus intacts en utilisant l’imagerie à montage entier, fournissant ainsi des informations sur le microenvironnement cellulaire de la graisse dans le muscle. De plus, ce protocole peut être combiné avec notre approche récemment publiée pour déterminer la section transversale des myofibres dans les tissus musculaires fixes31, une mesure importante pour évaluer la santé musculaire. La combinaison de cette approche avec le traçage de la lignée génétique pour cartographier le destin de la différenciation des FAP en adipocytes est également décrite ici. Ainsi, le protocole polyvalent décrit ici permet une évaluation rigoureuse et reproductible des PAF et de leur différenciation en graisse intramusculaire dans les coupes de tissus et les tissus intacts.
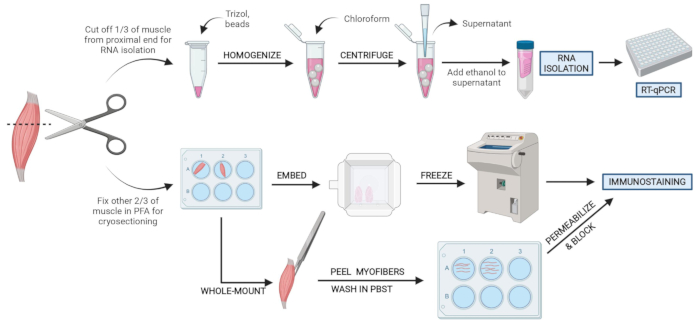
Figure 2 : Vue d’ensemble du protocole schématique. Vue d’ensemble schématique du traitement tissulaire dans lequel un tiers de l’AT est retiré, congelé et homogénéisé pour l’isolement ultérieur de l’ARN et l’analyse de transcription via RT-qPCR. Les deux autres tiers de l’AT sont fixés par PFA et traités pour l’immunocoloration sur des sections congelées ou des fibres à montage entier. Veuillez cliquer ici pour voir une version agrandie de cette figure.
Protocol
Representative Results
Discussion
Ce protocole décrit un protocole complet et détaillé qui permet une visualisation efficace et une quantification rigoureuse de la graisse intramusculaire. En divisant le même muscle en deux parties, l’une utilisée pour l’immunofluorescence et l’autre pour l’analyse RT-qPCR, ce protocole est également très polyvalent. Il peut également être combiné avec le traçage génétique de la lignée des FAP pour étudier leur conversion en adipocytes dans certaines conditions et est très adaptable pour étiqueter et imager plusieurs types de cellules supplémentaires.
Les moyens les plus couramment utilisés pour visualiser la graisse intramusculaire sont les sections de paraffine suivies de la coloration à l’hématoxyline et à l’éosine ou des sections congelées colorées pour les colorants lipophiles tels que l’huile rouge O (ORO). Cependant, alors que les tissus traités à la paraffine conservent la meilleure histologie, le même processus extrait également tous les lipides empêchant l’utilisation de colorants lipophiles. Bien que les méthodes de coloration lipophile fonctionnent à la fois sur les sections de tissus fixes et non fixées par PFA, les gouttelettes lipidiques sont facilement déplacées en appliquant une pression sur le couvercle, déformant ainsi la distribution spatiale de la graisse intramusculaire. Pour contourner ce problème, une étude récente a établi un protocole rigoureux pour visualiser les adipocytes ORO+ en utilisant une approche globale. Pour cela, les auteurs ont décellularisé l’AT pour visualiser la distribution spatiale de la graisse intramusculaire dans l’ensemble du TA41. Aussi puissante que soit cette technique, elle empêche également l’utilisation d’autres co-taches pour marquer des structures cellulaires supplémentaires. L’approche d’immunofluorescence globale présentée ici peut être utilisée pour co-colorer les adipocytes avec une variété de marqueurs permettant une cartographie fine de l’environnement cellulaire. Un défi majeur, cependant, est la pénétration tissulaire des anticorps. Plus les fibres sont maintenues ensemble, plus il sera difficile pour les anticorps de pénétrer et de lier également tous les antigènes disponibles. Ainsi, cette méthode est plus efficace lorsque l’on regarde de petits groupes de fibres. Dans le même temps, il s’agit également d’une limitation car l’emplacement anatomique global de la graisse intramusculaire est perdu en se concentrant uniquement sur de petits faisceaux de fibres décollées. Cependant, avec le développement actuel de nouvelles méthodes de nettoyage des tissus et de nouvelles technologies d’imagerie, une plus grande pénétration et visualisation des tissus sera possible à l’avenir 42,43,44.
Bien que la fixation préalable du tissu musculaire préserve la morphologie des adipocytes, il est également difficile d’évaluer la taille des myofibres, une mesure importante de la santé musculaire. La taille des myofibers est déterminée en mesurant la section transversale des myofibres. Nous avons déjà signalé qu’une fixation antérieure du tissu musculaire entraînera l’échec de la plupart des marqueurs disponibles pour décrire les myofibres31. Pour surmonter cet obstacle, nous avons développé un nouveau pipeline de segmentation d’image, qui permet de mesurer la taille du myofibre même dans les sections musculaires fixes31. Ainsi, nous avons établi un pipeline de traitement des tissus robuste et efficace qui, combiné à ce protocole, surmonte la plupart des inconvénients causés par la fixation préalable du tissu musculaire.
Un autre avantage majeur de cette approche est la polyvalence. En divisant l’AT en deux parties, la quantité d’informations pouvant être obtenues à partir d’un muscle est maximisée. Cela réduit non seulement le nombre d’animaux, mais ajoute également une couche supplémentaire de contrôle en confirmant l’histologie par l’expression des gènes et vice versa. En outre, de nombreux gènes différents peuvent être examinés au-delà des gènes adipogéniques. L’ARN isolé peut également être utilisé pour une expérience RNAseq musculaire entière. Enfin, le morceau musculaire surgelé peut également être utilisé pour le travail des protéines. L’une des limites de ce protocole est la possibilité que la blessure ne soit pas uniforme sur toute la longueur de l’AT. Cela pourrait conduire à un scénario où les deux parties musculaires divergent dans la quantité de graisse intramusculaire qu’elles contiennent et peut justifier l’exclusion d’un tel échantillon de toute analyse en aval. Il est donc recommandé de ne pas simplement s’appuyer sur la RT-qPCR pour tirer des conclusions majeures sur la quantité de graisse intramusculaire, mais plutôt comme données de soutien aux quantifications histologiques.
Ensemble, ce protocole décrit un pipeline de traitement tissulaire robuste, efficace et rigoureux qui permettra la visualisation et la quantification de la graisse intramusculaire, la première étape dans le développement de nouvelles options de traitement pour lutter contre la fibrose graisseuse. En même temps, il est polyvalent et peut être adapté à de nombreux types de cellules différentes dans le muscle ainsi que les adipocytes dans d’autres tissus.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Nous remercions les membres du laboratoire Kopinke d’avoir aidé à la collecte de données et à la lecture critique du manuscrit. Nous remercions également les membres de l’Institut de myologie de l’Université de Floride pour leur précieuse contribution au manuscrit. Le travail a été soutenu par la subvention NIH 1R01AR079449. La figure 2 a été créée avec Biorender.
Materials
| 16% PFA (Pack of 12, 10 mL bottles) | Electron Miscroscopy Sciences | 15710 | |
| 2.0 mL Microcentrifuge Tubes | Fisher Scientific | 05-408-138 | microcentrifuge tubes for snapfreezing/bead beating |
| 2-Methylbutane (4 L) | Fisher Chemical | O3551-4 | isopentane |
| Absolute Ethanol (200 proof) | ThermoFisher Scientific | BP2818100 | |
| AffiniPure Fab fragment donkey anti-mouse | Jackson ImmunoResearch | 715-007-003 | mouse-on-mouse blocking |
| Alexa Fluor 488 donkey anti-chicken secondary antibody | Jackson ImmunoResearch | 703-545-155 | |
| Alexa Fluor 488 donkey anti-mouse secondary antibody | Invitrogen | A21202 | |
| Alexa Fluor 488 donkey anti-rabbit secondary antibody | Invitrogen | A21206 | |
| Alexa Fluor 568 donkey anti-goat secondary antibody | Invitrogen | A11057 | |
| Alexa Fluor 568 donkey anti-rabbit secondary antibody | Invitrogen | A11037 | |
| Alexa Fluor 568 Phalloidin antibody | Invitrogen | A12380 | Dissolved in 1.5 mL methanol (~66 µM working solution) |
| BioLite 24-well Multidishes | ThermoFisher Scientific | 930186 | 24 well plate for PFA tissue incubation |
| Biometra TOne | analytikjena | 8462070301 | Thermal cycler |
| Chicken anti-GFP antibody | Aves Labs | GFP-1020 | |
| Chloroform/isoamyl alcohol 24:1(v/v) for molecular biology, DNAse, RNAse, and Protease free | ThermoFisher Scientific | AC327155000 | |
| Corn oil | Sigma Aldrich | C8267 | |
| DAPI stain | Invitrogen | D1306 | 150 µM working solution in dH2O |
| Donkey Serum (100 mL) | Millipore Sigma | 5058837 | for blocking solution |
| Dumont #5 Forceps | Fine Science Tools | 11251-20 | sharp-tipped tweezers |
| Fine Scissors Straight 9 cm | Fine Science Tools | 14060-09 | |
| Fluoromount-G | SouthernBiotech | 0100-01 | mounting medium |
| Glycerol, 99.5%, for molecular biology (500 mL) | Acros Organics | 327255000 | |
| Goat anti-PDGFRα antibody | R&D | AF1062 | |
| Hybridization Oven | VWR | 230301V | for Tamoxifen incubation |
| ImmEdge Hydrophobic Barrier PAP Pen | Vector Laboratories | H-4000 | hydrophobic pen |
| Insulin Syringe with Micro-Fine IV needle (28 G) | BD | 329461 | |
| Insulin Syringe with Slip Tip, 1 mL | BD | 329654 | Insulin syringe without needle, for oral gavaging |
| iScript cDNA Synthesis Kit | Bio-Rad | 1708890 | |
| Isoflurane | Patterson Veterinary | 78938441 | |
| Leica DMi8 inverted microscope | Leica | micrscope used for widefield IF and confocal imaging | |
| Micro Slides | VWR | 48311-703 | positively charged microscope slides |
| mouse anti-MYOD antibody | Invitrogen | MA1-41017 | |
| mouse anti-PAX7 antibody (supernatant) | DSHB | AB 428528 | |
| MX35 Premier+ Microtome blades | ThermoFisher Scientific | 3052835 | microtome blades |
| NanoDrop 2000 Spectrophotometer | ThermoFisher Scientific | ND2000 | spectrophotometer for RNA yield |
| Play-Doh | Hasbro | modeling compound | |
| PowerUp SYBR Green Master Mix | ThermoFisher Scientific | A25742 | green dye PCR master mix |
| Puralube Vet Ointment | Puralube | 17033-211-38 | vet ophthalmic ointment |
| QuantStudi 6 Flex Real-Time 384-well PCR System | Applied Biosystems | 4485694 | qPCR machine |
| Rabbit anti-perilipin antibody | Cell Signaling Technology | 9349S | |
| Red-Rotor Shaker | Hoefer Scientific | PR70-115V | shaker for IF staining |
| Richard-Allan Scientific Slip-Rite Cover Glass | ThermoFisher Scientific | 152460 | coverslips |
| RNeasy Mini Kit | QIAGEN | 74106 | contains mini spin columns |
| Safe-Lock Tubes 1.5 ml, natural | Eppendorf | 22363204 | |
| Sample Tubes RB (2 mL) | QIAGEN | 990381 | |
| Sodium azide | Alfa Aesar | 14314 | |
| Stainless Steel Beads, 2.8 mm | Precellys | KT03961-1-101.BK | small beads |
| Stainless Steel Beads, 5 mm | QIAGEN | 69989 | medium beads |
| Stainless Steel Beads, 7 mm | QIAGEN | 69990 | large beads |
| Stainless Steel Disposable Scalpels | Miltex | 327-4102 | scalpel |
| Stainless steel feeding tube, 20 G x 38 mm, straight | Instech Laboratories | FTSS-20S-3 | gavage needle |
| Tamoxifen | Toronto Research Chemicals | T006000 | |
| Tissue Plus O.C.T. Compound | Fisher HealthCare | 4585 | embedding medium |
| TissueLyser LT | QIAGEN | 85600 | bead beater |
| TissueLyser LT Adapter, 12-Tube | QIAGEN | 69980 | |
| Tissue-Tek Cryomold | Sakura | 4566 | specimen molds |
| Triton X-100 | Alfa Aesar | A16046 | |
| TRIzol Reagent | ThermoFisher Scientific | 15596026 | guanidium thiocyanate |
| Tween20 (500 mL) | Fisher BioReagents | BP337-500 | |
| VWR Micro Slides – Superfrost Plus | VWR | 48311703 | |
| Wheaton Coplin staining jars | Millipore Sigma | S6016 | Coplin jar |
References
- Milad, N., et al. Increased plasma lipid levels exacerbate muscle pathology in the mdx mouse model of Duchenne muscular dystrophy. Skeletal Muscle. 7 (1), 19 (2017).
- Goodpaster, B. H., et al. Obesity, regional body fat distribution, and the metabolic syndrome in older men and women. Archives of Internal Medicine. 165 (7), 777-783 (2005).
- Goodpaster, B. H., et al. Association between regional adipose tissue distribution and both type 2 diabetes and impaired glucose tolerance in elderly men and women. Diabetes Care. 26 (2), 372-379 (2003).
- Goodpaster, B. H., et al. The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. The Journals of Gerontology, Series A: Biological Sciences and Medical Sciences. 61 (10), 1059-1064 (2006).
- Goodpaster, B. H., Thaete, F. L., Kelley, D. E. Thigh adipose tissue distribution is associated with insulin resistance in obesity and in type 2 diabetes mellitus. American Journal of Clinical Nutrition. 71 (4), 885-892 (2000).
- Goodpaster, B. H., Theriault, R., Watkins, S. C., Kelley, D. E. Intramuscular lipid content is increased in obesity and decreased by weight loss. Metabolism. 49 (4), 467-472 (2000).
- Burakiewicz, J., et al. Quantifying fat replacement of muscle by quantitative MRI in muscular dystrophy. Journal of Neurology. 264 (10), 2053-2067 (2017).
- Murphy, W. A., Totty, W. G., Carroll, J. E. MRI of normal and pathologic skeletal muscle. American Journal of Roentgenology. 146 (3), 565-574 (1986).
- Willcocks, R. J., et al. Multicenter prospective longitudinal study of magnetic resonance biomarkers in a large duchenne muscular dystrophy cohort. Annals of Neurology. 79 (4), 535-547 (2016).
- Wokke, B. H., et al. Quantitative MRI and strength measurements in the assessment of muscle quality in Duchenne muscular dystrophy. Neuromuscular Disorders. 24 (5), 409-416 (2014).
- Contreras, O., Rossi, F. M. V., Theret, M. Origins, potency, and heterogeneity of skeletal muscle fibro-adipogenic progenitors-time for new definitions. Skeletal Muscle. 11 (1), 16 (2021).
- El Agha, E., et al. Mesenchymal stem cells in fibrotic disease. Cell Stem Cell. 21 (2), 166-177 (2017).
- Joe, A. W., et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nature Cell Biology. 12 (2), 153-163 (2010).
- Liu, W., Liu, Y., Lai, X., Kuang, S. Intramuscular adipose is derived from a non-Pax3 lineage and required for efficient regeneration of skeletal muscles. Developmental Biology. 361 (1), 27-38 (2012).
- Scott, R. W., Arostegui, M., Schweitzer, R., Rossi, F. M. V., Underhill, T. M. Hic1 defines quiescent mesenchymal progenitor subpopulations with distinct functions and fates in skeletal muscle regeneration. Cell Stem Cell. 25 (6), 797-813 (2019).
- Uezumi, A., et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. Journal of Cell Science. 124, 3654-3664 (2011).
- Hogarth, M. W., et al. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nature Communications. 10 (1), 2430 (2019).
- Uezumi, A., Fukada, S., Yamamoto, N., Takeda, S., Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nature Cell Biology. 12 (2), 143-152 (2010).
- Stumm, J., et al. Odd skipped-related 1 (Osr1) identifies muscle-interstitial fibro-adipogenic progenitors (FAPs) activated by acute injury. Stem Cell Research. 32, 8-16 (2018).
- Kopinke, D., Roberson, E. C., Reiter, J. F. Ciliary Hedgehog signaling restricts injury-induced adipogenesis. Cell. 170 (2), 340-351 (2017).
- Huang, Y., Das, A. K., Yang, Q. Y., Zhu, M. J., Du, M. Zfp423 promotes adipogenic differentiation of bovine stromal vascular cells. PLoS One. 7 (10), 47496 (2012).
- Sun, Y. -. M., et al. PDGFRα regulated by miR-34a and FoxO1 promotes adipogenesis in porcine intramuscular preadipocytes through Erk signaling pathway. International Journal of Molecular Sciences. 18 (11), 2424 (2017).
- Te, L. J. I., Doherty, C., Correa, J., Batt, J. Identification, isolation, and characterization of fibro-adipogenic progenitors (FAPs) and myogenic progenitors (MPs) in skeletal muscle in the rat. Journal of Visualized Experiments: JoVE. (172), e61750 (2021).
- Lukjanenko, L., et al. Aging disrupts muscle stem cell function by impairing matricellular WISP1 secretion from fibro-adipogenic progenitors. Cell Stem Cell. 24 (3), 433-446 (2019).
- Santini, M. P., et al. Tissue-resident PDGFRalpha(+) progenitor cells contribute to fibrosis versus healing in a context- and spatiotemporally dependent manner. Cell Reports. 30 (2), 555-570 (2020).
- Uezumi, A., et al. Identification and characterization of PDGFRalpha+ mesenchymal progenitors in human skeletal muscle. Cell Death & Disease. 5, 1186 (2014).
- Wosczyna, M. N., et al. Mesenchymal stromal cells are required for regeneration and homeostatic maintenance of skeletal muscle. Cell Reports. 27 (7), 2029-2035 (2019).
- Soliman, H., et al. Pathogenic potential of Hic1-expressing cardiac stromal progenitors. Cell Stem Cell. 26 (2), 205-220 (2020).
- Chung, M. I., Bujnis, M., Barkauskas, C. E., Kobayashi, Y., Hogan, B. L. M. Niche-mediated BMP/SMAD signaling regulates lung alveolar stem cell proliferation and differentiation. Development. 145 (9), (2018).
- Hilgendorf, K. I., et al. Omega-3 fatty acids activate ciliary FFAR4 to control adipogenesis. Cell. 179 (6), 1289-1305 (2019).
- Waisman, A., Norris, A. M., Elías Costa, M., Kopinke, D. Automatic and unbiased segmentation and quantification of myofibers in skeletal muscle. Scientific Reports. 11 (1), 11793 (2021).
- Kang, S. H., Fukaya, M., Yang, J. K., Rothstein, J. D., Bergles, D. E. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 68 (4), 668-681 (2010).
- Srinivas, S., et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Developmental Biology. 1, 4 (2001).
- Lukjanenko, L., Brachat, S., Pierrel, E., Lach-Trifilieff, E., Feige, J. N. Genomic profiling reveals that transient adipogenic activation is a hallmark of mouse models of skeletal muscle regeneration. PLoS One. 8 (8), 71084 (2013).
- Mahdy, M. A., Lei, H. Y., Wakamatsu, J., Hosaka, Y. Z., Nishimura, T. Comparative study of muscle regeneration following cardiotoxin and glycerol injury. Annals of Anatomy = Anatomischer Anzeiger: Official Organ of the Anatomische Gesellscaft. 202, 18-27 (2015).
- Pisani, D. F., Bottema, C. D., Butori, C., Dani, C., Dechesne, C. A. Mouse model of skeletal muscle adiposity: a glycerol treatment approach. Biochemical and Biophysical Research Communications. 396 (3), 767-773 (2010).
- Kawai, H., et al. Experimental glycerol myopathy: a histological study. Acta Neuropathologica. 80 (2), 192-197 (1990).
- Schmittgen, T. D., Livak, K. J. Analyzing real-time PCR data by the comparative CT method. Nature Protocols. 3 (6), 1101-1108 (2008).
- Uezumi, A., et al. Mesenchymal Bmp3b expression maintains skeletal muscle integrity and decreases in age-related sarcopenia. The Journal of Clinical Investigation. 131 (1), 139617 (2021).
- Biferali, B., et al. Prdm16-mediated H3K9 methylation controls fibro-adipogenic progenitors identity during skeletal muscle repair. Science Advances. 7 (23), 9371 (2021).
- Biltz, N. K., Meyer, G. A. A novel method for the quantification of fatty infiltration in skeletal muscle. Skeletal Muscle. 7 (1), (2017).
- Vieites-Prado, A., Renier, N. Tissue clearing and 3D imaging in developmental biology. Development. 148 (18), (2021).
- Gómez-Gaviro, M. V., Sanderson, D., Ripoll, J., Desco, M. Biomedical applications of tissue clearing and three-dimensional imaging in health and disease. iScience. 23 (8), 101432 (2020).
- Ueda, H. R., et al. Tissue clearing and its applications in neuroscience. Nature Reviews Neuroscience. 21 (2), 61-79 (2020).
