Quelle: Jonathan F. Blaize1, Elizabeth Suter1, und Christopher P. Corbo1
1 Department of Biological Sciences, Wagner College, 1 Campus Road, Staten Island NY, 10301
Die quantitative Bewertung von Prokaryoten kann angesichts ihres Überflusses, ihrer Neigung zur exponentiellen Proliferation, der Artenvielfalt innerhalb einer Population und der spezifischen physiologischen Bedürfnisse belastend sein. Diese Herausforderung verschlimmert sich in der vierphasigen Natur, in der sich Bakterien vermehren (Lag, Log, stationär und Tod). Die Fähigkeit, die Konzentration von Mikroorganismen genau abzuschätzen, ist für eine erfolgreiche Identifizierung, Isolierung, Kultivierung und Charakterisierung erforderlich (6). Daher haben Mikrobiologen seit über einem Jahrhundert serielle Verdünnungs- und verschiedene Beschichtungstechniken eingesetzt, um die bakterielle und virale Belastung in klinischen, industriellen, pharmazeutischen und akademischen Laborumgebungen zuverlässig zu quantifizieren (2,4,6). Beschreibungen dieser Methode erschienen erstmals 1883, als der deutsche Wissenschaftler und Arzt Robert Koch seine Arbeit über infektionserregende Erreger veröffentlichte (2). Kochs vorgenannte Techniken, die oft als Vater der modernen Bakteriologie bezeichnet werden, sind weltweit zum Goldstandard für die Aufzählung von Mikroorganismen geworden, die kultivierbar oder nicht mehr sind.
Serielle Verdünnung ist eine systematische Reduktion einer bekannten oder unbekannten Einheit (ein Gelöster, Organismus usw.) durch sukzessive Wiedersuspension einer Ausgangslösung (Lösung0) in feste Volumina eines flüssigen Verdünnungsmittels (Rohlinge). Diese Rohlinge bestehen in der Regel aus 0,45% Saline, obwohl die Zusammensetzung variiert werden kann (7). Während ein Experimentator ein beliebiges Volumen für jedes Verdünnungsmittel auswählen kann, ist es meistens ein Vielfaches von 10, was eine logarithmische Reduktion der Probe erleichtert. Beispielsweise enthält Lösung0 insgesamt 100 E. coli-Zellen, die in 10 ml Nährstoffbrühe suspendiert sind. Wenn 1 ml Lösung0 entfernt und zu 9 ml Saline (Verdünnungsstoff1)hinzugefügt wird, würde die neue Lösung (Lösung1) 1/10der Anfangskonzentration von E. colienthalten. In diesem Beispiel würde die neue Lösung (Lösung1) 10 E. coli-Zellen enthalten. Eine Wiederholung dieses Prozesses durch Entfernen von 1 ml Lösung1 und Hinzufügen zu weiteren 9 ml Saline (Verdünnungsmittel2) würde Lösung2ergeben, die nur eine einzelne E. coli-Zelle enthält. Da jede neue Lösung (9 ml Verdünnungsstoff + 1 ml Lösung) insgesamt 10 ml enthält, können wir schlussfolgern, dass der Verdünnungsfaktor für diese Reduktion 10 beträgt oder dass es sich um eine 10-fache serielle Verdünnung handelte (Abbildung 1). Da wir in diesem Beispiel erst mit 100 Zellen begonnen haben und wir um den Faktor 10 verdünnen, sind nur zwei Schritte erforderlich, um die absolute Mindestkonzentration von 1 Zelle zu erreichen.
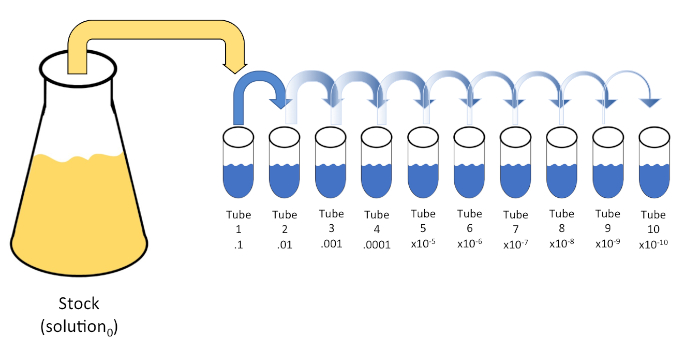
Abbildung 1: Serielle Verdünnung einer Lagerlösung. Ein 1 ml Aliquot der Stammlösung (Lösung0) wird zu Tube 1 hinzugefügt, die 9 ml mit 0,45% Salzsalze (dilent1)enthält; das Produkt dieser Mischung ist Lösung1. Wiederholen Sie dies, indem Sie 1 ml der neu erstellten Lösung1 aliquotieren und zu Tube 2 hinzufügen. Die Aliquotierung und Resuspension setzt sich auf diese Weise fort, bis die endgültige Röhre erreicht ist, wodurch die Lagerkonzentration mit jedem Schritt um den Faktor 10 verwässert wird. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Serielle Verdünnung ist die einfachste Technik zur Erzielung überschaubarer Konzentrationen eines gewünschten Organismus und wird durch Petrischalenstreifen und -streuungen ergänzt, nur zwei von vielen Beschichtungstechniken, die von Mikrobiologen verwendet werden. Dieser Vorteil dieses Ansatzes besteht darin, dass der Experimentator reine Stämme einer einzelnen Art oder separate Stämme von einer gemischten Population ernten kann (7). Streaking wird durch die Einführung eines Organismus in ein festes Medium (in der Regel bestehend aus Agarose) erreicht, auf dem er wachsen wird, wenn die entsprechenden Nährstoffe verfügbar sind. Das sanfte Schwenken einer sterilen Impfschleife über das Medium (so dass ein subtiler Streifen bleibt) in einem starren sinusförmigen Muster verteilt den Organismus proportional zur Frequenz der Wellenform des Experimentators. Die Aufteilung der Petrischale in Drittel oder Viertel (Quadrantstreifen) und die Verringerung der Häufigkeit jedes Streifens, wenn eine neue Region der Schale eingegeben wird, wird die Anzahl der Mikroorganismen, die diese Region besetzen können, allmählich reduzieren und einzelne Kolonien anstelle einer nicht quantifizierbaren bakteriellen Rasen. Die Streubeschichtung verdünnt die Proben nicht zusätzlich; ein steriler Glasstreuer wird verwendet, um ein Aliquot von Suspensionsmedien über eine ganze Petrischale zu verteilen (Abbildung 2). Die Kolonien, die auf der Streuplatte wachsen, entstehen aus einer einzigen Zelle und jede Kolonie auf der Schale kann gezählt werden, um die Anzahl der koloniebildenden Einheiten pro Milliliter (KBE) in einer gegebenen Suspension zu schätzen, dargestellt als KBE/ml (6) (Abbildung 3) Weicher Agar und Nachbau Beschichtungen sind Variationen der oben genannten Techniken und ermöglichen die Isolierung von Bakteriophagen bzw. Mutantenscreening (1,7).
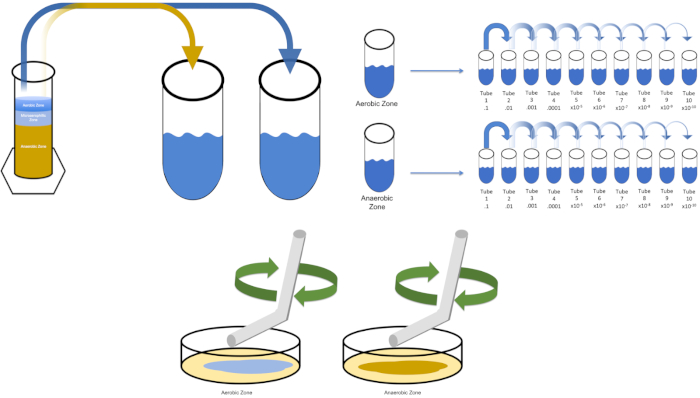
Abbildung 2: Plattenstreifen für bakterielle Aufzählung und Dehnungsisolierung. Beschriften Sie den Boden einer Petrischale mit Identifikationsinformationen (Bakterienname, Datum, Medien) und teilen Sie sie in Drittel auf. Nach der Auswahl einer geeigneten Verdünnung der Stammprobe eine sterile (Einweg- oder Flammen)-Impfschleife nehmen und das Reagenzglas untertauchen (hier, T3). Die Petrischalenabdeckung auf einer Seite leicht anheben, so dass nur die Impfschlaufe auf den Agar zugreifen kann. Gleiten Sie die Impfschleife über die Oberseite der Medien in einer Zick-Zack-Manier, um darauf zu achten, den Agar nicht zu kompromittieren. Drehen Sie die Platte um etwa 1/3rd (ca. 118°) und reduzieren Sie die Frequenz der Zick-Zack-Bewegung. Drehen Sie ein letztes Mal und reduzieren Sie die Zick-Zack-Frequenz noch einmal. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
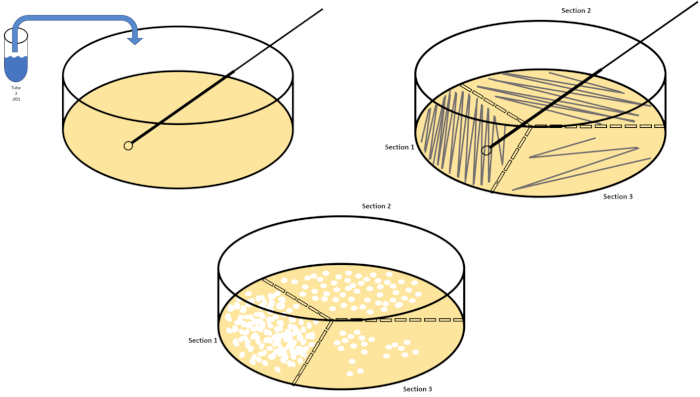
Abbildung 3: Streubeschichtung. 1 g der Aerobic-Zone wurde in T1 resuspendiert und dann seriell verdünnt. Ein steriler Glas- oder Kunststoff-Einweg-Streustab wird verwendet, um Inoculum in jeder Schale zu verteilen. Dies wurde mit 1 g der anaeroben Zone wiederholt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Wie bei seriellen Verdünnungen wird eine logarithmische Skala verwendet, um die Konzentration des Organismus auszudrücken. Die Anzahl der Kolonien, die in Standard-Petrischalen mit einer Größe von 100 mm x 15 mm angebaut werden, kann manuell (oder automatisiert mit Hilfe der Computerverarbeitung) aufgezählt werden, indem isolierte Wachstumscluster identifiziert werden. Zählt, die insgesamt weniger als 30 oder mehr als 300 wert sind, sollten als zu wenig zum Zählen (TFTC) oder zu zahlreich für die Anzahl (TNTC) definiert werden. Im letzteren Fall sollte eine serielle Verdünnung durchgeführt werden, um die Konzentration zu reduzieren, bevor eine neue Petrischale wieder aufgelegt wird. Die Mittelung der Anzahl der in sich geschlossenen Kolonien, die aus drei getrennten Petrischalen identifiziert wurden, und multiplizierte den Mittelwert mit dem Verdünnungsfaktor ergibt CFU/ml; Das Zeichnen des Protokolls10 von KFU/ml gegen die Zeit wird die mittlere Erzeugungszeit des Organismus offenbaren (7).