Fuente: Jonathan F. Blaize1, Elizabeth Suter1y Christopher P. Corbo1
1 Departamento de Ciencias Biológicas, Wagner College, 1 Campus Road, Staten Island NY, 10301
La evaluación cuantitativa de los prokaryotes puede ser onerosa dada su abundancia, propensión a la proliferación exponencial, diversidad de especies dentro de una población y necesidades fisiológicas específicas. Acomenda este desafío, está la naturaleza de cuatro fases en la que las bacterias se replican (retraso, registro, estacionario y muerte). La capacidad de estimar con precisión la concentración de microorganismos es necesaria para una identificación, aislamiento, cultivo y caracterización exitosas (6). Como tal, los microbiólogos han empleado la dilución en serie y diversas técnicas de chapado durante más de un siglo para cuantificar de forma fiable la carga bacteriana y viral en entornos clínicos, industriales, farmacéuticos y académicos de laboratorio (2,4,6). Las descripciones de esta metodología aparecieron por primera vez en 1883 cuando el científico y médico alemán Robert Koch publicó su trabajo sobre agentes causantes de enfermedades infecciosas (2). A menudo conocido como el padre de la bacteriología moderna, las técnicas de Koch se han convertido en el estándar de oro para la enumeración de microorganismos, cultivables o de otro tipo, en todo el mundo.
La dilución en serie es una reducción sistemática de una entidad conocida o desconocida (un soluto, organismo, etc.) mediante la resuspensión sucesiva de una solución inicial (solución0)en volúmenes fijos de un diluyente líquido (en blanco). Estos espacios en blanco suelen consistir en 0,45% de salina, aunque la composición puede ser variada (7). Mientras que un experimentador puede elegir cualquier volumen para cada diluyente, más a menudo es un múltiplo de 10, facilitando la reducción logarítmica de la muestra. Por ejemplo, la solución0 contiene un total de 100 células de E. coli suspendidas en 10 ml de caldo nutritivo. Si se retira 1 ml de la solución0 y se añade a 9 ml de solución salina (diluyente1), la nueva solución (solución1) contendría 1/10de la concentración inicial de E. coli. En este ejemplo, la nueva solución (solución1) contendría 10 células de E. coli. Repetir este proceso eliminando 1 ml de la solución1 y añadiéndola a otros 9 ml de solución salina (diluyente2)produciría la solución2,que contiene sólo una sola célula de E. coli. Dado que cada nueva solución (9 ml de diluyente + 1 ml de solución) contiene un total de 10 ml, podemos concluir que el factor de dilución para esta reducción es de 10 o que se trataba de una dilución serial de 10 veces(Figura 1). Puesto que sólo comenzamos con 100 células en este ejemplo y nos estamos diluyendo por un factor de 10, sólo se requieren dos pasos para alcanzar la concentración mínima absoluta de 1 célula.
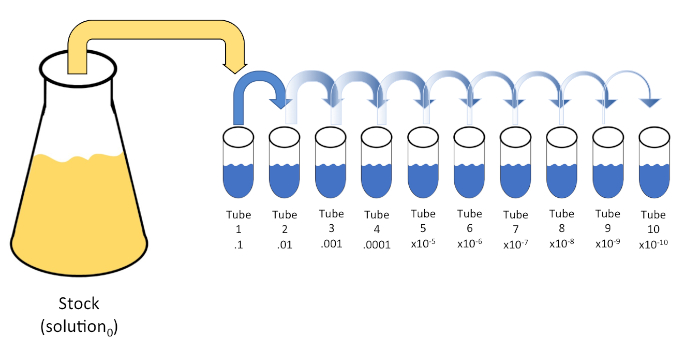
Figura 1: Dilución en serie de una solución de stock. Se añade una alícuota de 1 ml de la solución en stock (solución0) al tubo 1 que contiene 9 ml de solución salina al 0,45% (dilent1); el producto de esta mezcla es la solución1. Repita alístique1 ml de la solución recién creada1 y agregándola al tubo 2. La alícuota y la resuspensión continúan de esta manera hasta que se alcanza el tubo final, diluyendo la concentración de stock por un factor de 10 cada uno con cada paso. Haga clic aquí para ver una versión más grande de esta figura.
La dilución en serie es la técnica más sencilla para obtener concentraciones manejables de un organismo deseado y se complementa con el rayado y la propagación de la placa de petri, sólo dos de las muchas técnicas de chapado utilizadas por los microbiólogos. Este beneficio de este enfoque es que el experimentador puede cosechar cepas puras de una sola especie o cepas separadas de una población mixta (7). El rayado se logra introduciendo un organismo en un medio sólido (generalmente consistente en agarosa) sobre el que crecerá si se dispone de los nutrientes adecuados. Barrer suavemente un bucle inoculante estéril a través del medio (de modo que quede una racha sutil) en un patrón sinusoidal rígido distribuirá el organismo proporcionalmente a la frecuencia de la forma de onda del experimentador. Dividir la placa Petri en tercios o cuartos (racha de cuadrante) y disminuir la frecuencia de cada racha a medida que se introduce una nueva región del plato reducirá gradualmente el número de microorganismos que pueden ocupar esa región, produciendo colonias únicas en lugar de una césped bacteriano incuantificable. El revestimiento extendido no diluye además las muestras; un esparcidor de vidrio estéril se utiliza para distribuir una alícuota de medios de suspensión a través de un plato de petri completo(Figura 2). Las colonias que crecen en la placa de propagación surgen de una sola célula y cada colonia en el plato se puede contar para estimar el número de unidades formadoras de colonias por mililitro (CFU) en una suspensión dada, representada como CFU/mL (6)(Figura 3) Agar suave y réplica El revestimiento son variaciones de las técnicas antes mencionadas y permiten el aislamiento de bacteriófagos y cribado mutante, respectivamente (1,7).
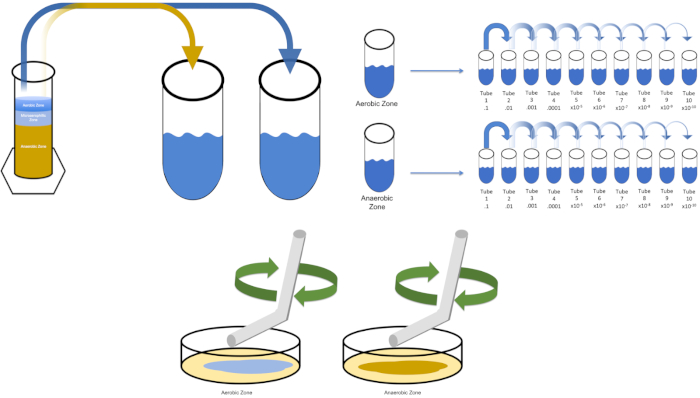
Figura 2: Rayado de placas para la enumeración bacteriana y aislamiento de deformación unitaria. Etiquete la parte inferior de una placa de petri con información de identificación (nombre de bacteria, fecha, medios) y divídala en tercios. Después de seleccionar una dilución adecuada de la muestra de stock, tome un bucle de inoculado estéril (desechable o flameado) y sumerja el tubo de ensayo (aquí, T3). Levante ligeramente la cubierta de la placa de petri en un lado para que sólo el bucle inoculado pueda acceder al agar. Deslice el bucle inoculante a través de la parte superior de los medios de comunicación de una manera en zig-zag teniendo cuidado de no comprometer el agar. Gire la placa aproximadamente 1/3rd (118o) y reduzca la frecuencia del movimiento en zig-zag. Gire una última vez y reduzca la frecuencia en zig-zag una vez más. Haga clic aquí para ver una versión más grande de esta figura.
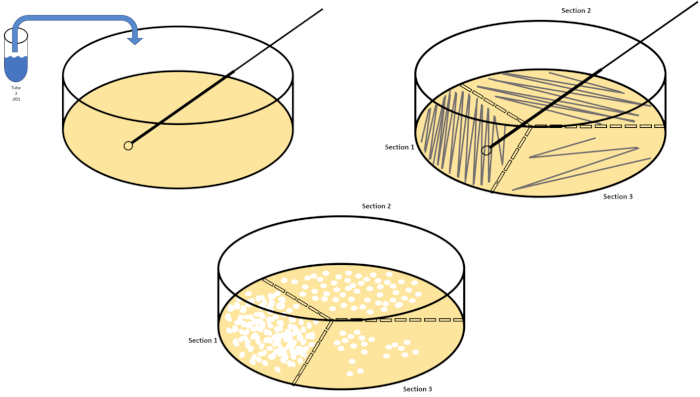
Figura 3: Extender el revestimiento. 1 g de la zona aeróbica se resuspendió en T1 y luego se diluyó en serie. Para distribuir el inóculo por cada plato se utiliza una varilla de esparcimiento desechable de vidrio estéril o plástico para distribuir el inóculo. Esto se repitió con 1 g de la zona anaeróbica. Haga clic aquí para ver una versión más grande de esta figura.
Al igual que con las diluciones en serie, se emplea una escala logarítmica para expresar la concentración del organismo. El número de colonias cultivadas en platos de petri estándar de 100 mm x 15 mm se puede enumerar manualmente (o automatizar con la ayuda del procesamiento computacional) mediante la identificación de racimos aislados de crecimiento. Cuenta que el total de menos de 30 o más de 300 debe definirse como demasiado pocos para contar (TFTC) o demasiado numerosos para contar (TNTC), respectivamente. En el caso de este último, se debe realizar una dilución en serie para reducir la concentración antes de volver a rayar una nueva placa de petri. Promediar el número de colonias autónomas identificadas a partir de tres platos de petri separados y multiplicar la media por el factor de dilución producirá CFU/ml; trazando el registro10 de CFU/ml contra el tiempo revelará el tiempo medio de generación del organismo (7).