Identification of Orexin and Endocannabinoid Receptors in Adult Zebrafish Using Immunoperoxidase and Immunofluorescence Methods
Summary
Presented here are protocols for immunohistochemical characterization and localization of orexin peptide, orexin receptors, and endocannabinoid receptors in the gut and brains of normal and diet-induced obesity (DIO) adult zebrafish models using immunoperixidase and double immunofluorescence methods.
Abstract
Immunohistochemistry (IHC) is a highly sensitive and specific technique involved in the detection of target antigens in tissue sections with labeled antibodies. It is a multistep process in which the optimization of each step is crucial to obtain the optimum specific signal. Through IHC, the distribution and localization of specific biomarkers can be detected, revealing information on evolutionary conservation. Moreover, IHC allows for the understanding of expression and distribution changes of biomarkers in pathological conditions, such as obesity. IHC, mainly the immunofluorescence technique, can be used in adult zebrafish to detect the organization and distribution of phylogenetically conserved molecules, but a standard IHC protocol is not estasblished. Orexin and endocannabinoid are two highly conserved systems involved in the control of food intake and obesity pathology. Reported here are protocols used to obtain information about orexin peptide (OXA), orexin receptor (OX-2R), and cannabinoid receptor (CB1R) localization and distribution in the gut and brain of normal and diet-induced obese (DIO) adult zebrafish models. Also described are methods for immunoperoxidase and double immunofluorescence, as well as preparation of reagents, fixation, paraffin-embedding, and cryoprotection of zebrafish tissue and preparation for an endogenous activity-blocking step and background counterstaining. The complete set of parameters is obtained from previous IHC experiments, through which we have shown how immunofluorescence can help with the understanding of OXs, OX-2R, and CB1R distribution, localization, and conservation of expression in adult zebrafish tissues. The resulting images with highly specific signal intensity led to the confirmation that zebrafish are suitable animal models for immunohistochemical studies of distribution, localization, and evolutionary conservation of specific biomarkers in physiological and pathological conditions. The protocols presented here are recommended for IHC experiments in adult zebrafish.
Introduction
Immunohistochemistry (IHC) is a well-established classic technique used to identify cellular or tissue components (antigens) by antigen-antibody interaction1,2. It can be used to identify the localization and distribution of target biomolecules within a tissue. IHC uses immunological and chemical reactions to detect antigens in tissue sections3. The main markers used for the visualization of antigen-antibody interactions include fluorescent dyes (immunofluorescence) and enzyme-substrate color reactions (immunoperoxidase), both conjugated to antibodies4. Using microscopic observation is possible to determine the localization of labeled tissue, which approximately corresponds to localization of the target antigen in the tissue.
Two methods exist for fluorescent or chromogenic reactions to detect protein: the direct detection method, in which the specific primary antibody is directly labeled; and the indirect detection method, in which the primary antibody is unconjugated while the secondary antibody carries the label5,6,7. The indirect method has some advantages, which is mainly its signal amplification. Moreover, unlike other molecular and cellular techniques, with immunofluorescence, it is possible to visualize the distribution, localization, and coexpression of two or more proteins differentially expressed within cells and tissues7. The choice of the detection method used depends on experimental details.
To date, IHC is widely used in basic research as a powerful and essential tool to understanding the distribution and localization of biomarkers and the general profiling of different proteins in biological tissue from human to invertebrates8,9,10,11. The technique helps display a map of protein expression in a large number of normal and altered animal organs and different tissue types, showing possible down- or up-regulation of expression induced by physiological and pathological changes. IHC is a highly sensitive technique that requires accuracy and the correct choice of methods to obtain optimal results12. First of all, many different factors such as fixation, cross-reactivity, antigen retrieval, and sensitivity of antibodies can lead to false positive and false negative signals13. Selection of the antibodies is one of the most important steps in IHC and depends on the antigen specificity and its affinity to the protein and species under investigation7.
Recently, we have optimized the IHC technique to detect members of orexin/hypocretin and endocannabinoid systems in adult zebrafish tissue. We have focused mainly on fixation, tissue embedding using two different approaches, sectioning and mounting (which can affect resolution and detail during microscopic analysis), and blocking (to prevent false positives and reduce background)14. Other important characteristics are the antibody specificity and selectivity and reproducibility of individual IHC protocols. The key to providing antibody specificity is the use of negative controls (including no primary antibodies or tissue that is known to not express the target proteins) as well as positive controls (including tissue that is known to express the target proteins)15. The selection of antibodies for IHC is made based on their species-specificity (the likelihood with which they react with the antigen of interest) and the antigen-antibody binding detection systems that is used4,5,6,7. In the case of immunoperoxidase, the color of the reaction is determined by selection of the precipitating chromogen, usually diaminobenzidine (brown)16. On the other hand, immunofluorescence utilizes antibodies conjugated with a fluorophor to visualize protein expression in frozen tissue sections and allows for easy analysis of multiple proteins with respect to the chromogenic detection system5,7.
In the immunoperoxidase technique, the secondary antibody is conjugated to biotin, a linker molecule capable of recruiting a chromogenic reporter molecule [avidin-biotin complex (ABC)], leading to amplification of the staining signal. With the ABC reporter method, the enzyme peroxidase reacts with 3,3’-diaminobenzidine (DAB), producing an intensely brown-colored staining where the enzyme binds to the secondary antibody, which can then be analyzed with an ordinary light microscope. ABC staining, due to the high affinity of avidin for biotin, produces a rapid and optimal reaction, with few secondary antibodies attached to the site of the primary antibody reactivity. This chromogenic detection method allows for the densitometric analysis of the signal, providing semi-quantitative data based on the correlation of brown signal levels with protein expression levels18.
With immunofluorescence techniques, simultaneous detection of multiple proteins is possible due to the ability of different fluorochromes to emit light at unique wavelengths, but is important to choose fluorochromes carefully to minimize spectral overlap5. Moreover, the use of primary antibodies in different host species minimizes difficulties concerning cross-reactivity. In this case, each species-specific secondary antibody recognizes only one type of primary antibody. Fluorescent reporters are small organic molecules, including commercial derivatives, such as Alexa Fluor dyes.
Many animal models are used to understand particular physiological and pathological conditions. To date, it is established that many metabolic pathways are conserved over the course of evolution. Therefore, IHC studies in model organisms such as zebrafish can provide insight into the genesis and maintenance of pathological and non-pathological conditions17. It is an aim of this report to illustrate IHC protocols that can be performed on adult zebrafish tissue and used to obtain detailed images of the distribution and localization of OXA, OX-2R, and CB1R at peripheral and central levels. Also reported are protocols for the application of two major IHC indirect methods in peripheral and central tissues of adult zebrafish. Described is the indirect method, which allows for signal amplification in cases where a secondary antibody is conjugated to a fluorescent dye (immunofluorescence method) or enzyme reporter (immunoperoxidase method). Both chromogenic and fluorescent detection methods possess advantages and disadvantages. Reported in this protocol is the use of IHC, mainly immunofluorescence, in adult zebrafish, an animal model widely used to study systems that are evolutionary conserved across different physiological and pathological conditions.
Protocol
1. Immunoperoxidase protocol
NOTE: The zebrafish were obtained by Prof. Omid Safari (Department of Fisheries, Faculty of Natural Resources and Environment, Ferdowsi University of Mashhad, Mashhad, Iran)10.
- Tissue dissection
- Sacrifice the zebrafish by submersion in ice water (5 parts ice/1 part water, 4 °C); leave them until cessation of all movement to ensure death by hypoxia.
- Quickly remove the gut and brain with the following dissection method:
- Dry the fish on a paper towel and place it sagittally on a dissecting mat, blocking the ventral part of the eye socket and fleshy part of the tail.
- For the intestine: cut the skin and carefully remove the skin and underlying muscle from the side of the fish until the internal organs are visible. Remove the intestine from the body cavity stretching it out.
- For the brain: remove the head with a razor blade. Remove soft tissue from the ventral side of the skull with forceps. Open the skull and remove the bone from the ventral side of the brain. Remove the skin and skull bones from the dorsal side of the brain. Remove the brain.
- Tissue fixation
- Fix the dissecting tissues by immersion in fresh 4% paraformaldeyde (PFA) in phosphate buffer (PB, pH 7.4, 4 °C) for 3 h at room temperature (RT).
- Prepare 0.1 M PB by dissolving 1.755 g of NaH2PO4H2O and 4.575 g of Na2HPO4 in 450 mL of distilled water (dH2O), and adjust to a pH of 7.4 with the necessary amount of NaOH 1N.
- Prepare 4% PFA dissolving 4 g of PFA in 100 mL of 0.1 M PB (pH 7.4) by agitation on a heat plate. When a temperature of 60 °C is reached, add 2-4 drops of NaOH 1N to obtain a clear solution. Left PFA 4% at RT and control the pH . Adjust the pH to 7.4.
NOTE: Tissue fixation for more than 4–6 h may lead to overfixation, which masks antigens, limiting antibody-epitope binding. Time of fixation depends on tissue size.
- Fix the dissecting tissues by immersion in fresh 4% paraformaldeyde (PFA) in phosphate buffer (PB, pH 7.4, 4 °C) for 3 h at room temperature (RT).
- Tissue embedding
- Rinse the tissues 5x for 5 min each, by immersion in 0.1 M PB (pH 7.4).
- Dehydrate the tissues by subsequent immersion in alcohol 70% (6 min), 80% (6 min), 95% (5 min), 95% (5 min), 100% (1 min), and 100% (1 min).
- Clarify the tissues by immersion in 100% alcohol:xylene (1:1) for 10 min, then 2x in xylene for 5 min each.
- Infiltrate the tissues with paraffin wax (56 °C) twice for 1 h each by direct immersion in paraffin.
- Embed the tissues in paraffin blocks at room temperature (RT) and store at RT until sectioning.
- Tissue cutting
- Cut tissues into coronal or sagittal sections with 8 µm thickness with a microtome, and collect the sections in alternate series onto adhesive glass slides on the water and warmed at 38 °C.
- Dry the slides with sections at 37 °C overnight.
- Deparaffinization and rehydratation of tissue sections
- Dewax the sections by immersion in xylene for 5 min followed by immersion in xylene for 3 min.
- Rehydrate the sections by subsequent slides immersion in alcohol 100% II (1 min), 100% I (1 min), 95% (1 min), 75% (1 min), 50% (1 min), then place the slides in dH2O (5 min).
NOTE: Completely dewax the sections before starting the reaction. If the sections still have traces of paraffin, perform an additional immersion in xylene for 5 minutes or more.
- Antigen retrieval
- Treat the sections with citrate buffer (0.01 M, pH 6.0) by immersing the slides in the solution and heating in a microwave oven for 5 min at maximal power.
- Let the sections cool.
- Repeat the cycle of 5 min in the microwave at maximal power to complete the antigen retrieval.
- Prepare citrate buffer (0.01 M, pH 6.0) by dissolving 2.10 g of citric acid in 100 mL of dH2O and 5.882 g of sodium citrate in 200 mL of dH2O. Mix sodium citrate (0.01 M) with citric acid (0.01 M) at a 1:4 ratio by volume and adjust the pH to 6 using HCl 0.1N.
- Blocking endogenous peroxidase
- Demarcate the tissue area on the slides with a solvent-resistant pen.
- Rinse the sections 3 times, 5 min each, with tris-buffered saline solution (TBS) (0.1 M, pH 7.3).
- Prepare 0.1 M TBS by dissolving 12.1 g of trizma base and 9 g of NaCl in 950 mL of dH2O and adjust to pH 7.3 with HCl 0.1N.
- Block the endogenous peroxidase activity by slides immersion in a solution of 0.75% H2O2 for 5 min at RT.
- Prepare the 0.75% H2O2 solution dissolving 5 mL of 30% H2O2 in 195 mL of dH2O.
NOTE: ndogenous enzymes can react with IHC reagents and yield false positive results. Moreover, highly vascularized tissues express many endogenous peroxidase, which can lead to intense nonspecific staining and background levels. Treatment with 0.75% H2O2 quenches endogenous peroxidase and significantly reduces the nonspecific background.
- Prepare the 0.75% H2O2 solution dissolving 5 mL of 30% H2O2 in 195 mL of dH2O.
- Blocking of nonspecific binding sites and tissue permeabilization
- Incubate the tissue sections with the TBS/0.4% Triton (TBS-T) blocking buffer, containing the primary antiserum (NRS), for 30 min at RT to block nonspecific binding sites.
- Prepare 0.4% Triton by dissolving 0.4 mL of Triton X-100 in 100 mL of TBS.
- Prepare the blocking buffer TBS-T solution by dissolving 1% normal serum in TBS containing 0.4% Triton X-100 (1 mL of normal serum + 8.2 mL of TBS + 0.8 mL of 0.48 Triton X-100).
NOTE: Select the species of the animal serum depending on the host of the secondary antibody (e.g., when using a goat anti-rabbit secondary antibody, use normal goat serum).
- Incubate the tissue sections with the TBS/0.4% Triton (TBS-T) blocking buffer, containing the primary antiserum (NRS), for 30 min at RT to block nonspecific binding sites.
- Tissue incubation with primary antibody
- Incubate the sections overnight in a humid box at RT with primary antibodies diluted in TBS-T. Stain the sections with the following primary antibody:
- Goat antibody against OX-2R diluted 1:200, which recognizes the C-terminal of the protein.
- Goat antibody against OX-A diluted 1:200 [orexin-A (C-19)], which recognizes the C-terminal of the protein.
NOTE: Ensure that the primary antibody reacts with the biomolecule of interest. Define with which epitope of the biomolecule the primary antibody reacts by using the gene alignment. For instance, the OX-A epitope has been mapped between the aa 50–100 of human OX-A (O43612) whereas the OX-2R epitope has been mapped near the last 50 aa at the C-terminal of human.
- Incubate the sections overnight in a humid box at RT with primary antibodies diluted in TBS-T. Stain the sections with the following primary antibody:
- Tissue incubation with secondary antibody
- Rinse the sections 3x for 5 min each, with 0.1 M TBS (pH 7.3).
- Incubate the sections for 1.5 h at RT with biotinylated secondary antibody and rabbit anti-goat immunoglobulin (H+L) conjugated with biotin, then dilute 1:100 in TBS-T.
NOTE: Test and find the different dilutions of both primary and secondary antibodies to find the optimal dilution that allows reduction of the background.
- Peroxidase reaction
- Rinse the sections 3x for 5 min each, with 0.1 M TBS (pH 7.3).
- Incubate the sections with avidin-biotin complex (ABC) for 1 h.
- Prepare ABC solution adding two drops of component A followed by two drops of component B in 5 mL of TBS.
- Rinse the sections 3x for 5 min each, with 0.1 M TBS (pH 7.3).
- Treat the sections with the chromogen substrate 3,3’-diaminobenzidine-4 (DAB).
- Prepare DAB solution adding one drop (approximately 30 µL) of DAB chromogen concentrate to 1 mL of DAB diluent, and mix well before use.
NOTE: Prepare ABC solution at least 30 min before use and keep the complex at 4 °C until use to allow for stable avidin/biotin binding. Prepare the fresh DAB working solution, apply to the tissue sections, and develop until the color is appropriate. When the chromogenic reaction converts the epitope sites to a brown color and the intensity of the signal is appropriate for imaging, proceed to the next step. Timing of DAB development may vary from a few seconds to 10 min. For OX-A and OX-2R 1, 2 min is enough. Handle DAB with care, as it is carcinogenic.
- Prepare DAB solution adding one drop (approximately 30 µL) of DAB chromogen concentrate to 1 mL of DAB diluent, and mix well before use.
- Tissue dehydratation and mounting
- Rinse all the sections 3x for 5 min each, with 0.1 M TBS (pH 7.3) to stop the DAB reaction.
- Dehydrate the sections by subsequent slides immersion in alcohol 50% (2 min), 75% (2 min), 95% (2 min), 100% I (2 min), 100% II (2 min).
- Clarify the slices by immersion 2x in xylene 10 min each.
- Mount the sections with DPX (dibutyl phthalate xylene) mountant for histology, adding two drops of mounting media to slides and topping slowly with coverslips.
NOTE: Since the immersion of tissue in increasing concentrations of alcohol during dehydration results in alcohol penetration of tissue and replacement of water with alcohol, determine a precise dehydration time and do not over dehydrate the tissues. Perform the immersion in xylene after dehydration to clarify the tissue and remove any excess or remaining alcohol.
- Controls
- Repeat sections 1.1–1.12, omitting the primary antibody and/or substituting the primary antibody or the secondary antibody IgG by TBS (negative controls).
- Repeat sections 1.1–1.12 pre-absorbing each primary antibody with an excess of the relative peptide (100 mg of peptide/1 mL of diluted antiserum).
- Repeat sections 1.1–1.12 using different tissue as positive control (e.g., mouse tissue).
NOTE: Prepare all buffers fresh, shortly before starting. Carefully remove the excess fluid at each step with a pipette or filter paper around the sections, always keeping the sections wet.
- Image acquisition and analysis
- Acquire digital images under constant light illumination and at the same magnification using a microscope equipped with a digital camera.
- Perform a semi-quantitative analysis of staining intensity using imaging software (see Table of Materials).
- Open the captured images, in .lif or .tiff file format, for evaluating indices of OX-A or OX2-R positivity.
- Select the button under Measure and click on Manual Counting. Count the number of stained cell profiles directly from the screen by placing a mark onto positive cells by clicking the mouse.
- Select a region of interest (ROI) area (3 x 103 μm2) to quantify the immunosignal density (optical density, OD).
- Determine the pixel with the highest intensity (high positive) and least intensity (negative) inside the analyzed image to assign the zero as the value of the background (i.e., a portion of tissue devoid of stained cells).
- Assess the OD by working on a logarithmic scale of absorbance. In digital image analysis, the DAB pixel intensity ranges from the darkest (0 value) to lightest (255 value) shade.
- Determine OD by plotting a histogram of the following: log10(255/I), where “I” is the pixel intensity value given by the program and determined by subtracting the background.
NOTE: The permanent and stable immunoperoxidase reaction can be analyzed under a bright-field microscope at any time. Perform the counting of labeled cells on alternate section to avoid double-counting of the same cell from adjacent sections.
2. Immunofluorescence protocol
- Tissue dissection
- Sacrifice and dissect the animals as described in section 1.1.
- Tissue fixation
- Fix the gut and brain by immersion for 3 h in 4% PFA at 4 °C.
- Prepare PB and 4% PFA as in section 1.2.1.
NOTE: The time of fixation depends on tissue size. Tissue fixation for more than 4–6 h may lead to overfixation, which masks antigens and limits antibody-epitope binding.
- Prepare PB and 4% PFA as in section 1.2.1.
- Fix the gut and brain by immersion for 3 h in 4% PFA at 4 °C.
- Tissue embedding
- Rinse the tissues 3x for 5 min each, with 0.1 M PB (pH 7.4).
- For the cryoprotection, transfer the tissues to 20% sucrose in PB (0.1 M, pH 7.4) and keep overnight at 4 °C. Then, transfer the tissues to 30% sucrose in 0.1 M PB (pH 7.4) and keep for an additional night at 4 °C.
- Embed the tissues in a block of optimal cutting temperature (OCT) compound. To do this, prepare a small dewar of liquid nitrogen, take aluminum foil, and cut it in half to create a pan. The pan containing the tissues filled with the OCT compound has to be dipped in the liquid nitrogen until the OCT compound is cooled. The frozen tissues can be stored at -80 °C until sectioning.
- Tissue cutting
- Transfer the frozen tissue blocks to a cryostat at -20 °C and cut in coronal or sagittal sections of 10 μm.
- Collect the tissues in alternate serial sections onto adhesive glass slides suitable for immunohistochemistry and store them at -20 °C until use.
NOTE: Store slides between -20 °C and 4 °C in a dark slide box or slide book.
- Blocking of nonspecific binding sites and tissue permeabilizzation
- Demarcate the tissue area on the slide with a solvent resistant pen.
- Rinse the sections 3x for 5 min each, with 0.1 M PB (pH 7.4).
- Incubate the sections with 1% normal donkey serum dissolved in the permeabilization buffer PB-Triton X-100 0.3% (PB-T) for 30 min at RT to permeabilize the cell membrane and block the nonspecific binding sites.
- Prepare Triton X-100 0.3% dissolving 0.3 mL of Triton X-100 in 100 mL of 0.1 M PB (pH 7.4).
- Prepare the blocking solution dissolving 1% normal donkey serum in PB containing 0.3% TritonX-100.
NOTE: The animal species of the serum used in the permeabilization and blocking buffers are dependent on the host of the secondary antibody.
- Incubation with mix of primary antibodies
- Rinse the sections 3x for 5 min each, with 0.1 M PB (pH 7.4).
- Incubate the sections overnight in a humid box at RT with a mix of primary antibodies diluted in PB-T. The following mixes of primary antibodies can be used: goat antibody against OX-2R diluted 1:100/rabbit antibody against CB1R diluted 1:100, or goat antibody against OX-A diluted 1:100/rabbit antibody against CB1R diluted 1:100.
- Rinse the sections 3x for 5 min each, with 0.1 M PB (pH 7.4).
- Incubation with mix of secondary antibodies
- Rinse the sections 3x for 5 min each, with 0.1 M PB (pH 7.4).
- Incubate the sections for 2 h at RT with 1) a mix of donkey anti-rabbit Alexa Fluor 488-conjugated secondary antibody and donkey anti-goat Alexa Fluor 594-conjugated secondary antibody diluted 1:100 in PB-T; or 2) a mix of donkey anti-goat Alexa Fluor 488-conjugated secondary antibody and donkey anti-rabbit Alexa Fluor 594-conjugated secondary antibody diluted 1:100 in PB-T.
NOTE: Use secondary antibodies developed in the same animal host. The normal serum must belong to the same species of the secondary antibody (e.g., use secondary antibodies developed in donkey and a donkey normal serum). Dilute the primary and secondary antibodies with blocking buffer containing the detergent to increase cell permeabilization and reduce background.
- Tissue mounting
- Rinse the sections 3x for 5 min each, with 0.1 M PB (pH 7.4).
- Counterstain the sections with nuclear dye DAPI (4’,6-diamidino-2-phenylindole) prepared dissolving 1.5 µL of DAPI (1 mg/mL) in 3 mL of PB.
- Coverslip slides with mounting medium (see Table of Materials). This aqueous mounting medium stabilizes the tissue sample and stains for long-term usage. Fluorescent samples can be stored in the dark at 4 °C. To prolong the life of fluorofores, use an antifed mounting medium.
NOTE: Choose a good mounting medium. One of the most important parameters of mounting agents is the refractive index (nD), which should be around 1.5, the refractive index of glass. The mounting medium used here can be used especially with specimen prepared for enzyme and lipid determinations (i.e., specimen that must not be dehydrated with an ascending series of alcohol).
- Controls
- Repeat sections 2.1–2.8, omitting the primary or secondary antibody, or substituting in the specific step the primary or secondary antisera with PB (negative control).
- Repeat sections 2.1–2.8, pre-absorbing each primary antibody with an excess of the relative peptide (100 mg of peptide/1 mL of diluted antiserum).
- Repeat sections 2.1–2.8 on slices of mouse brain (positive control).
NOTE: Prepare all the buffers fresh, shortly before starting. Remove the excess of fluid carefully at each step with a pipette or filter paper around the sections, always keeping the section humid.
- Image acquisition
- Use a confocal microscope equipped with an x-y-z motorized stage, digital camera, and acquisition and image analysis software such as NIS-Elements C to observe and analyze the immunostained sections. Acquire digital images using the 5-20-40x objectives.
- Take the images of each section at low magnification (10x or 20x objective) in each of the available channels to compose a low magnification montage, providing an overview of the entire region to facilitate the localization and documentation of OX-2R/CB1R anatomical co-expression. Normalize the fluorescence images to maximum contrast and overlay before the analysis.
- Collect serial Z-stacks of images throughout the area of interest. Acquire the images through six focal planes (Z-step) with focal steps of 1–1.8 µm to cover the tissue volume in which OX-2R/CB1R coexpression is visualized as yellow puncta. To do this collection for each channel (red, green, blue) separately, by using a Z-motorized microscope.
- Use an imaging deconvolution software to deconvolve images by application of ten iterations and collapse the serial Z planes images into a single maximum projection image.
- Adjust the micrographs for light and contrast using Adobe Photoshop 6.01 (Adobe Systems, San Jose, CA).
NOTE: Perform the deconvolution step during observation to further decrease the background. Limit the slide exposure to light to prevent photobleaching.
- Image analysis
- Perform quantitative analysis of OX-2R/CB1R coexpression on alternated 10 μm thick sections (n = 5 animal per group) by covering all the region of interest of each animal.
- Quantify the OX-2R/CB1R coexpression as number of yellow positive puncta with a semiautomated system of image analysis. Imagins software can provide a higher level of detail, with quantitative data regarding the regions of overlap between different fluorescent probes.
- Quantify the puncta by using thresholding tools for signal intensity in the two channels. Open the .liff, .tiff, or .jpg images file and select Image–Threshold. Select Auto Setting or Manual Method and regulate Threshold until all the stained puncta are selected. Analyze the distribution of the pixel intensities in an area of the image that does not contain any immunolabelled objects to obtain the background threshold. Determine this background individually for every image. The program then removes the background threshold by setting the baseline of pixel intensities to the background value.
- Select Analyze–Measure and choose the parameters to be measured (puncta intensity-minimum, maximum and mean values; number/density of the immunolabelled puncta).
- Count the yellow puncta along the volume of the tissue in 4 Z-stacks for each section, using the stacks immediately above or below to the best focus plane. Exclude the out-of-focus regions from the analysis.
NOTE: Perform the counting of labeled cells on alternate section to avoid double-counting of the same cells from adjacents sections.
Representative Results
Representative data for the immunoperoxidase staining are shown in Figure 1 and Figure 2. Immunohistochemical analysis of OX-A and OX-2R distribution in the gut of adult zebrafish showed different localization sites of OX-A and OX-2R and their increases in expression in the intestinal cells of DIO zebrafish. An intense brown staining for OX-A was observed in the cells of the medial and anterior intestine (Figure 1A, A1). The immunoexpression of OX-A gave clear signals in the different gut compartements, decreasing from the anterior toward the medial intestine (Figure 1B, B1). The negative control was used as a reference for the background and to confirm the specificity of OX-A signal (Figure 1E). The prolonged exposure to the chromogen DAB resulted in the increase of the background intensity (Figure 1D). Similar results were observed for OX-2R immunoexpression in the intestine of DIO and control diet zebrafish (Figure 2). An increased OX-A signal in DIO adult zebrafish was accompanied by the overexpression of OX-2R in others intestinal compartements (Figure 2B, B1).
Using immunofluorescence, the data obtained by immunoperoxidase analysis were confirmed, underlying the increase of OX-A and OX-2R expression in the gut of adult DIO zebrafish with respect to the control (Figure 3). Moreover, using double immunofluorescence, it was possible to obtain information about the expression of the endocannabinoid receptor CB1R and its co-localization with OX-A or OX-2R in the gut and brain of control diet and DIO adult zebrafish. The advantage of immunofluorescence is that it yields a more detailed signal with less provided information about tissue morphology, compared to immunoperoxidase technique. Using the immunofluorescence method, we have previously determined anatomical interactions between OX-2R and CB1R in both the gut and brain10.
The accurate analysis of immunofluorescent images showed the increase of OX-2R/CB1R co-localization in the gut of DIO adult zebrafish compared to the control diet zebrafish (Figure 3B, C). A similar situation was observed in different brain regions, such as the dorsal telencephalon, hypothalamus (lateral, ventral, and dorsal zones), optic tectum, torus lateralis, and diffuse nucleus of the inferior lobe (Figure 4). The negative (Figure 5A,B) and positive (mouse brain) (Figure 5C) controls were used as references for the background and to confirm the specificity of CB1R and OX-2R signals.
Moreover, by double immunostaining with OX-A/CB1R, it was observed in the orexinergic neurons of the hypothalamus that there was an increase of co-localization accompanied by an increase of OX-A fluorescent signal (Figure 6). These results show how double immunofluorescence can help to identify physiologically conserved protein expression, co-localization of target proteins, and their distribution and/or expression changes in different pathological conditions.
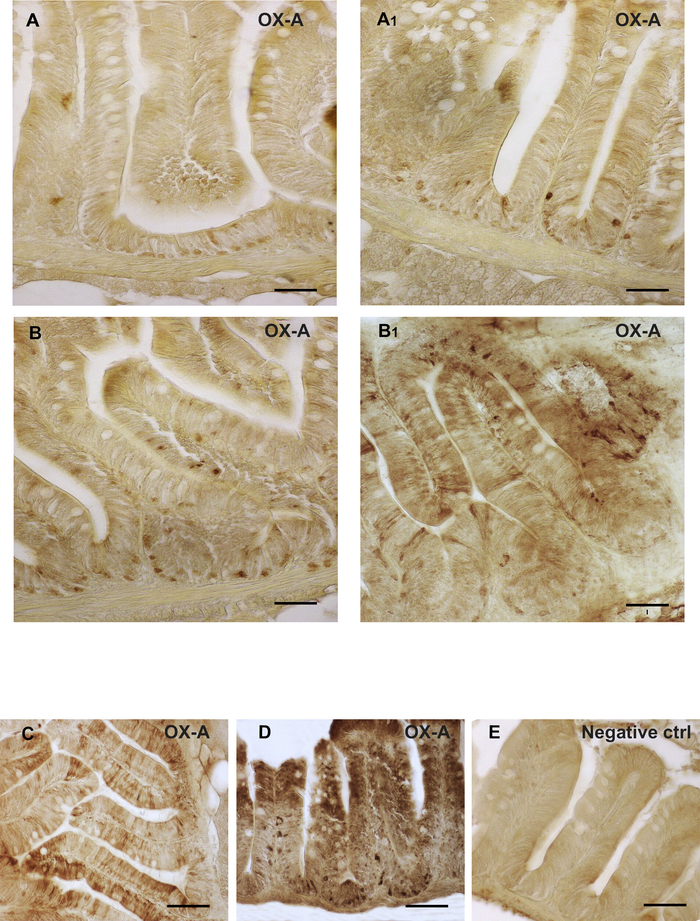
Figure 1: Orexin immunolocalization in the intestines of DIO vs. control diet adult zebrafish. (A) OX-A immunoreactivity in the cells of the medial intestine of control diet zebrafish. (A1) OX-A immunoreactivity in the cells of the medial intestine of DIO zebrafish. (B) OX-A immunoreactivity in the cells of the anterior intestine of control diet zebrafish. (B1) OX-A immunoreactivity in the cells of the anterior intestine of DIO zebrafish. (A1, B1) An increase of OX-A positive cells in different tissue compartement of the medial and anterior intestine of DIO zebrafish. (C) OX-A immunoreactivity in the cells of the anterior intestine of DIO zebrafish. (D) immunoperoxidase reaction for OX-A after a prolonged exposure to DAB. (E) Negative control. Scale bar: 50 µm (A, A1, B, B1); 100 µm (C, D, E). Please click here to view a larger version of this figure.
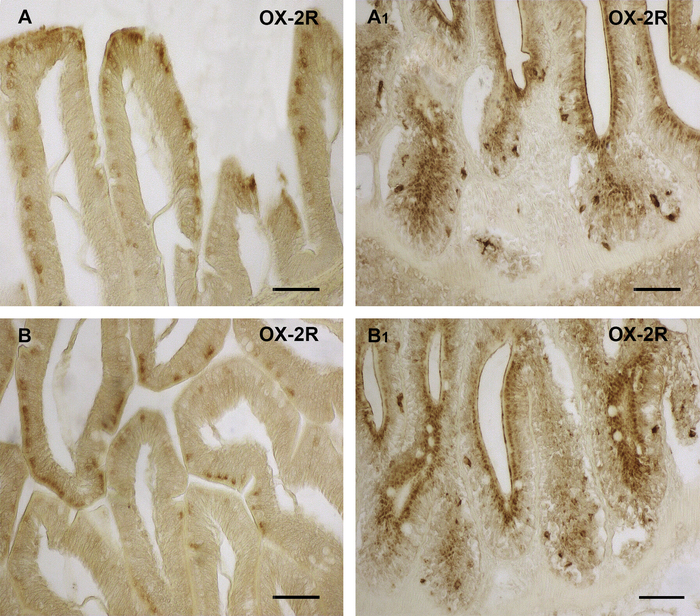
Figure 2: Orexin 2 receptor immunolocalization in the intestine of DIO vs. control diet adult zebrafish. (A) OX-2R immunoreactivity in the cells of the medial intestine of control diet zebrafish. (A1) OX-2R immunoreactivity in the cells of the medial intestine of DIO zebrafish. (B) OX-2R immunoreactivity in the cells of the anterior intestine of control diet zebrafish. (B1) OX-2R immunoreactivity in the cells of the anterior intestine of DIO zebrafish. (A1, B1) An increase of OX-2R positive cells in different tissue compartement of the medial and anterior intestine of DIO zebrafish. Scale bar: 50 µm. Please click here to view a larger version of this figure.
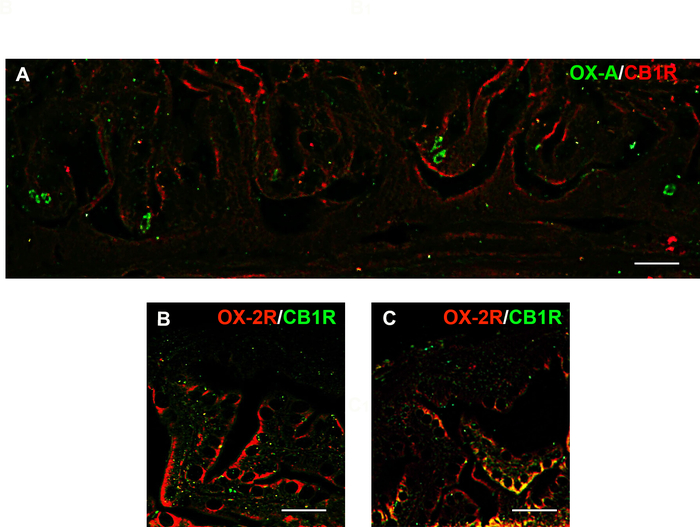
Figure 3: Distribution of OX-A (green)/CB1R (red) and OX-2R (red)/CB1R (green) and their co-localization with OX-2R/CB1R (yellow) in the intestines of DIO vs. control diet adult zebrafish. (A) OX-A/CB1R co-expression in the intestine of control diet adult zebrafish. (B) OX-2R/CB1R co-expression within the intestine of control diet zebrafish. (C) OX-2R/CB1R co-expression within the intestine of DIO adult zebrafish. An increase of OX-2R/CB1R co-localization (yellow dots) in the intestine of DIO zebrafish. Scale bar: 25 µm. Please click here to view a larger version of this figure.
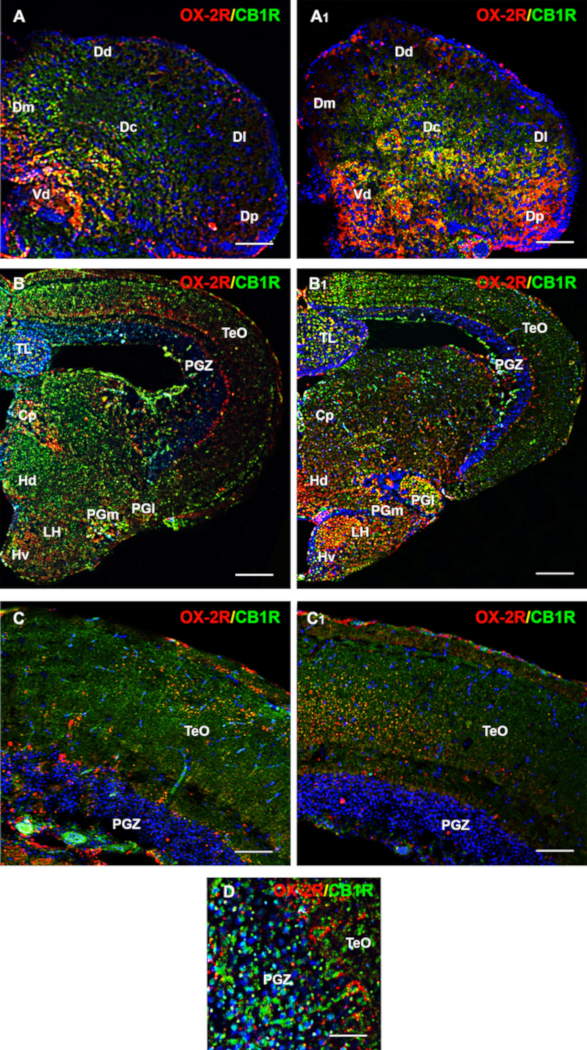
Figure 4: Distribution of OX-2R (red) and CB1R (green) and their co-localization with OX-2R/CB1R (yellow) in the brain coronal sections of DIO vs. control diet adult zebrafish. (A) OX-2R/CB1R co-expression in the telencephalon of the control diet zebrafish. (A1) OX-2R/CB1R co-expression in the telencephalon of DIO zebrafish. (B) OX-2R/CB1R co-expression within the lateral, ventral and dorsal zone of hypothalamus, optic tectum, torus lateralis, diffuse nucleus of the inferior lobe of control diet zebrafish (B1) OX-2R/CB1R co-expression within the lateral, ventral, and dorsal zones of the hypothalamus, optic tectum, torus lateralis, and diffuse nucleus of the inferior lobe of DIO zebrafish. (C) Higher magnification of the optic tectum showing the co-expression of OX-2R/CB1R (yellow) in the control diet zebrafish. (C1) Higher magnification of the optic tectum showing the co-expression of OX-2R/CB1R (yellow) in the DIO zebrafish. (D) A particular of the optic tectum showing the distribution and co-expression of OX-2R/CB1R (yellow) in the DIO zebrafish. DAPI (blue) was used to counterstain nuclei. CP: central posterior thalamic nucleus; Dd: dorsal; Dc: central; Dl: lateral; Dm: medial; Dp: posterior part of the dorsal telencephalon; Hd: dorsal zone of the periventricular hypothalamus; Hv: ventral zone of the periventricular hypothalamus; LH: lateral part of the hypothalamus; PGl: lateral and PGm: medial preglomerular nuclei; PGZ: periventricular gray zone of the optic tectum; TeO: optic tectum; TL: torus longitudinalis; Vd: dorsal part of the ventral telencephalon. Scale bar: 50 µm (A, A1, C, C1); 250 µm (B and B1); 25 µm (D). Please click here to view a larger version of this figure.
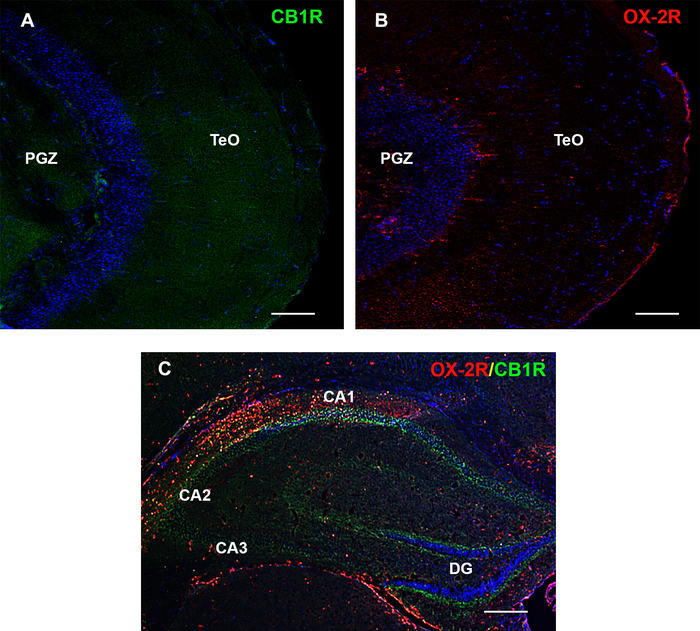
Figure 5: OX-2R and CB1R protein expression and specificity in adult brain of zebrafish and mouse hippocampus. (A) Negative control of OX-2R by pre-absorption with the relative peptide. (B) Negative control of CB1R by pre-absorption with the relative peptide. (C) Positive control of OX-2R/CB1R in hippocampus of mouse. PGZ: periventricular grey zone of the optic tectum; TeO: optic tectum. Scale bar: 100 µm (A, B); 250 µm (C). Please click here to view a larger version of this figure.
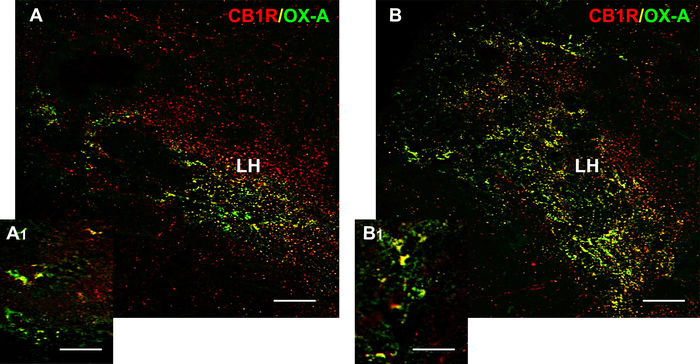
Figure 6: Distribution of OX-A (green) and CB1R (red) and their co-localization with OX-A/CB1R (yellow) in the hypothalamic coronal sections of DIO vs. control diet adult zebrafish. (A) OX-A/CB1R co-expression in the hypothalamus of control diet zebrafish (A1) Higher magnification showing the OX-A/CB1R co-expression within the lateral hypothalamus. Detail of OX-A/CB1R co-expression showing a putative adjacent localization of OX-A/CB1R or co-localization and overlap of OX-A/CB1R in the same cells. (B) OX-A/CB1R co-expression in the Hypothalamus of DIO zebrafish (B1) Higher magnification showing the increased OX-A/CB1R co-expression within the lateral hypothalamus. Detail of OX-A/CB1R co-expression showing a putative adjacent localization of OX-A/CB1R or co-localization and overlap of OX-A/CB1R in the same cells. LH: lateral part of the hypothalamus. Scale bar: 50 µm (A, B); 25 µm (A1, B1). Please click here to view a larger version of this figure.
Discussion
Sample preparation
Sample preparation is the first critical step in IHC. A reliable protocol allows for maintenance of cell morphology, tissue architecture, and antigenicity. This step requires correct tissue collection, fixation, and sectioning22,23. The purpose of fixation is to preserve tissue and reduce the action of tissue enzymes or microorganisms. In particular, the fixation step preserves cellular components and biomolecules, prevents autolysis and shifting of cell constituents (such as antigens and enzymes), stabilizes cellular materials against aversive effects of the following procedures, and facilitates immunostaining4,7,24.
Before sectioning, the tissue is prepared and preserved through paraffin embedding (immunoperoxidase method) or cryopreserved (freezing in cryomedia, in multiple immunofluorescence methods). The preservation method is associated with the type of fixation7. After fixation and preservation, the tissues are sliced by a microtome if embedded in paraffin, or by a cryostat if embedded in a cryomedia. Tissues are typically sliced at a thickness range of 8-10 μm and mounted on slides. For immunoperoxidase staining, the sample may require additional steps to unmask the epitopes for antibody binding, including deparaffinization and antigen retrieveal25,26. It should be kept in mind that overfixation can cause epitope masking, while underfixation can cause little to no positive signal with heavy edge staining.
Blocking and background reduction
In the immunoperoxidase method, the high affinity of avidin for biotin is likely responsible for the rapid production of background staining. Moreover, since the avidin-biotin reaction is irreversible, the background cannot be removed18,27. During IHC, high background can also be produced by nonspecific binding to endogenous tissue biotines. Hydrophobic and ionic interactions (such as those produced by collagen and other connective tissues such as epithelium and adipocytes), as well as endogenous enzyme activity, are major causes of background staining. Endogenous biotin or enzymes and hydrophobic binding must be minimized prior to antibody staining, which can be achieved by the addition of a detergent, such as Triton X-100, in the blocking buffers28.
The use of 0.3%-0.4% Triton X-100 in the blocking buffers also allows for full permeabilization of the antibodies into tissue sections. Moreover, although antibodies are preferentially specific for one epitope, partial binding to sites on nonspecific proteins is possible, leading to high background staining29. The nonspecific bindings can mask the signal of the target antigen30. To reduce nonspecific background staining, samples should be incubated with a buffer that blocks nonspecific reactive sites. Common blocking buffers include normal serum or bovine serum albumin30. The species of the blocking serum should be the same as the host of the secondary antibody. It is recommended to determine the best incubation time. Concentration of the normal serum in the blocking buffer is another important determination31. Furthermore, to eliminate the background staining, it is crucial to use the optimal dilution of the primary and secondary antibodies. Thus, incubation time must be chosen carefully, in addition to the temperature (i.e., increase the time if performing at 4 °C, decrease the time if at RT) and detection system.
Antibody choice
The selection of antibodies for IHC staining is important and can affect experimental outcome32. To ensure that the antibody will respond appropriately, the epitope recognized by it must be considered. Understanding the target protein and its function, tissue and subcellular localization, and whether it undergoes post-translational modifications can help to determine the choice of antibody. Another important step is the testing of different concentrations of primary/secondary antibody to keep the background and aspecificity at a minimum level compatible with a specific signal. Moreover, it is important to check species reactivity to confirm primary and secondary antibody compatibility and the capability of the primary antibody to recognize the antigen target in its native conformation. Another important step for primary antibody choice is gene alignment. This ensures that the primary antibody reacts with the biomolecule of interest and provides information about which epitope is recognized by the antibody in a specific animal model. Gene alignment also provides the possibility of choosing antibodies capable of recognizing epitopes that are evolutionary conserved.
Controls
To clarify specificity of the antibodies, an important aspect is performance of the controls, which allows detection of specific staining. The controls include: i) a tissue known to express the antigen as a positive control; ii) a tissue known not to express the antigen as a negative control; iii) the omission of the primary antibody or absorption of the primary antibody with a specific peptide to confirm that the secondary antibody does not crossreact with other tissue components14,33.
In IHC techniques, several steps can cause problems prior to achieving the final staining. Strong background staining and nonspecific target antigen staining can be caused by endogenous biotin or primary/secondary antibody cross-reactivity and poor enzyme activity or primary antibody potency, respectively. Background staining and nonspecific binding can be prevented by blocking the endogenous enzymes prior to the incubation with the primary antibody. Using normal serum is the best way to block nonspecific interactions30. The choice of the blocking buffer depends on the method of detection used. Moreover, a tissue known to lack the expression of the target antigen as negative control can act as the reference to determine the amount of background staining34. The deposition of chromogenic or fluorescent signal in the negative control confirms the presence of nonspecific staining. Furthermore, insufficient blocking time blocking leads to a high background, while excessive blocking leads to a low signal35.
In immunoperoxidase staining, the presence of endogenous peroxidase in the tissue is another cause of brown background deposition. The treatment with H2O2 prior the incubation with the primary antibody blocks the endogenous peroxidase36,37. On the contrary, in immunofluorescence, fixation plays a key role in the generation of autofluorescence. To avoid autofluorescence, the best fixation method, time of fixation, and preparation of tissues must be carefully chosen. Another critical step of IHC is validation of the primary antibody. An antibody is considered valid if it produces a consistent and specific staining pattern in a particular tissue or cell/subcellular components and if pre-absorption of the primary antibody with a specific peptide does not yield staining38. In immunofluorescence, nonspecific binding shows similar fluorescent intensity under three color fluorescence detectors, while the signal is variable since different fluorescent conjugated secondary antibodies are used. These aspects of IHC tissue preparation and antibody staining must be addressed to overcome staining issues.
IHC, although a relatively simple technique, presents some limitations and depend on many factors39. One of the crucial points is formalin fixation, which can alter the expression of post-translation modified proteins. On the other hand, formalin-fixed paraffin-embedded tissues can be stored for long-term at room temperature, whereas frozen tissues can only be stored for up to one year at -80 °C. Moreover, frozen tissues can be damaged by the formation of ice crystals, which can affect the subcellular details altering IHC staining40,41. Regarding our studies, the most difficult aspect was finding specific primary antibodies against zebrafish molecules. Although zebrafish have been recognized as a valid animal model with a highly conserved degree of structure, very few antibodies have been developed that can recognize specific proteins and other molecules in zebrafish. To overcome these limitations, it is important to validate antibody specificity by western blotting analysis and gene alignment.
Growing interest in IHC methods has led to the development of highly specific immunostaining that can help investigative studies42. IHC is being used with increasing frequency to identify the presence of specific molecular markers and their changes across different pathologies. The two approaches illustrated here, immunoperoxidase and immunofluorescence, have respective advantages and disadvantages43. Paraffin-embedded tissue, used in the immunoperoxidase technique, can allow for high resolution of cells and tissue and reveal details about the distribution and amount of target proteins44,45. However, paraffin-embedded tissues are not suitable for immunofluorescence, since paraffin can mask the antigenicity and lead to nonspecific fluorescence46. On the other hand, cryosection of PFA-fixed tissue preserves endogenous antigenicity and lead to a decrease in nonspecific fluorescence. Even if the technical quality of the cryosections are much lower than that of paraffin-embedded tissue, they can yield valid results using the IHC technique47.
Moreover, while paraffin embedding better preserves morphological details, cryopreservation better preserves enzyme and antigen expression, leading to more detailed immunostaining. The immunofluorescence technique also allows the contemporary detection of two or three different biomolecules, revealing possible interactions, as illustrated in our previous work for orexin and endocannabinoid systems10. Among the techniques used to detect the distribution and levels of specific biomolecules, IHC allows not only the determination of specific morphological expression and distribution of molecules and proteins, but also the possibility to perform quantitative analysis.
Using immunofluorescence, the codistribution and coexpression of biomolecules can also be further understood, as well as possible interactions and their changes in cases of different pathologies48. Confocal microscopy, during the last 20 years, has often been used to study the cellular and subcellular distribution of numerous proteins in the mammalian brain. Fluorescence microscopy also allows the visualization (using fluorophores or fluorescent dyes) of specific structures of interest, such as proteins, organelles, and other biological matter; such signals can be used in both fixed and living biological systems to image specific subcellular structures49. The potential modulatory function of biomolecules in specific cell compartments can be explored via the visualization of their expression patterns, whereas the role of protein signaling within the tissues may be uncovered via the neuroanatomical distribution of metabolic enzyme expression or receptors.
The presented technical work introduces IHC approaches, mainly immunofluorescence, to studying two highly conserved systems, orexin and endocannabinoid, in an adult zebrafish model. In particular, immunofluorescence methods can be used to determine the distribution, relative amount, and anatomical interactions of specific proteins in target tissues. Cryopreservation of PFA-fixed tissues better preserves highly sensitive proteins susceptible to rapid deterioration. Moreover, cryopreservation is thought to better preserve antigen and antigenicity, and it allows for studying of post-translation modified proteins and DNA.
Even if frozen tissues (compared to paraffin-embedded sections) are thicker, which hampers the ability to observe tissue morphology in detail, confocal microscopy allows for sample visualization in great detail and enhances imaging capabilities. Moreover, immunofluorescence can be used for quantitative analysis of staining intensity, and densitometric analysis of the signal provides quantitative data. This allows for determining correlations of fluorochrom signal levels with protein expression levels by looking at areas of co-localization. This work describes and illustrates protocols that can be used to study evolutionary conservation of important proteins in the adult zebrafish. The use of IHC, mainly immunofluorescence, in adult zebrafish can help highlight the usefulness of this animal model in studying the morphological expression and distribution of highly conserved biomolecules, as well as reveal possible alterations across different pathological conditions correlating with human physiopathology.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This study was supported by Fondi Ricerca di Ateneo (FRA)2015-2016, University of Sannio.
Materials
| Anti CB1 | Abcam | ab23703 | |
| Anti OX-2R | Santa Cruz | sc-8074 | |
| Anti-OXA | Santa Cruz | sc8070 | |
| Aquatex | Merck | 1,085,620,050 | |
| Biotinylated rabbit anti-goat | Vector Lab | BA-5000 | |
| citric acid | Sigma Aldrich | 251275 | |
| Confocal microscope | Nikon | Nikon Eclipse Ti2 | |
| Cryostat | Leica Biosystem | CM3050S | |
| DAPI | Sigma Aldrich | 32670 | |
| Digital Camera | Leica Biosystem | DFC320 | |
| Digital Camera for confocal microscope | Nikon | DS-Qi2 | |
| Donkey anti goat Alexa fluor 488-conjugated secondary antibodies | Thermo Fisher | A11055 | |
| Donkey anti goat Alexa fluor 594-conjugated secondary antibodies | Thermo Fisher | A11058 | |
| Donkey anti rabbit Alexa fluor 488-conjugated secondary antibodies | Thermo Fisher | A21206 | |
| Donkey anti rabbit Alexa fluor 594-conjugated secondary antibodies | Thermo Fisher | A21207 | |
| Ethanol absolute | VWR | 20,821,330 | |
| Frozen section compound | Leica Biosystem | FSC 22 Frozen Section Media | |
| H2O2 | Sigma Aldrich | 31642 | |
| HCl | VWR | 20,252,290 | |
| ImmPACT DAB | Vector lab | SK4105 | |
| Microscope | Leica Biosystem | DMI6000 | |
| Microtome | Leica Biosystem | RM2125RT | |
| Na2HPO4 | Sigma Aldrich | S9763 | |
| NaCl | Sigma Aldrich | S7653 | |
| NaH2PO4H2O | Sigma Aldrich | S9638 | |
| NaOH | Sigma Aldrich | S8045 | |
| Normal Donkey Serum | Sigma Aldrich | D9663 | |
| Normal Rabbit Serum | Vector lab | S-5000 | |
| paraffin wax | Carlo Erba | 46793801 | |
| Paraphormaldeyde | Sigma Aldrich | P6148 | |
| sodium citrate dihydrate | Sigma Aldrich | W302600 | |
| Triton X-100 | Fluka Analytical | 93420 | |
| Trizma | Sigma Aldrich | T1503 | |
| VectaStain Elite ABC Kit | Vector lab | PK6100 | |
| Xylene Pure | Carlo Erba | 392603 |
References
- Brandtzaeg, P. The increasing power of immunohistochemistry and immunocytochemistry. Journal of Immunological Methods. 216, 49-67 (1998).
- Haines, D. M., West, K. H. Immunohistochemistry: forging the links between immunology and pathology. Veterinary Immunology and Immunopathology. , 151-156 (2005).
- Onul, A., et al. Application of immunohistochemical staining to detect antigen destruction as a measure of tissue damage. Journal of Histochemistry & Cytochemistry. 60 (9), 683-693 (2012).
- Ramos-Vara, J. A. Technical aspects of immunohistochemistry. Veterinary Pathology. 42 (4), 405-426 (2005).
- Coons, A. H., Kaplan, M. H. Localization of antigen in tissue cells, II: improvements in a method for the detection of antigen by means of fluorescent antibody. The Journal of Experimental Medicine. 91 (1), 1-13 (1950).
- Coons, A. H., Leduc, E. H., Connolly, J. M. Studies on antibody production, I: a method for the histochemical demonstration of specific antibody and its application to a study of the hyperimmune rabbit. The Journal of Experimental Medicine. 102 (1), 49-60 (1955).
- Ramos-Vara, J. A., Miller, M. A., et al. When tissue antigens and antibodies get along: revisiting the technical aspects of immunohistochemistry–the red, brown, and blue technique. Veterinary Pathology. 51 (1), 42-87 (2014).
- Duraiyan, J., Govindarajan, R., Kaliyappan, K., Palanisamy, M. Applications of immunohistochemistry. Journal of Pharmacy and Bioallied Sciences. 2 (Suppl 2), S307-S309 (2012).
- Al-Hussinee, L., et al. Immunohistochemistry and pathology of multiple Great Lakes fish from mortality events associated with viral hemorrhagic septicemia virus type IVb. Diseases of Aquatic Organisms. 93 (2), 117-127 (2011).
- Imperatore, R., et al. Overlapping Distribution of Orexin and Endocannabinoid Receptors and Their Functional Interaction in the Brain of Adult Zebrafish. Frontiers in Neuroanatomy. 12, 62 (2018).
- Concas, A., et al. Immunochemical Localization of GABAA Receptor Subunits in the Freshwater Polyp Hydra vulgaris. Neurochemical Research. 41 (11), 2914-2922 (2016).
- Mania, M., et al. Expression and distribution of leptin and its receptors in the digestive tract of DIO (diet-induced obese) zebrafish. Annals of Anatomy. , 37-47 (2017).
- Matos, L. L., Trufelli, D. C., de Matos, M. G., da Silva Pinhal, M. A. Immunohistochemistry as an important tool in biomarkers detection and clinical practice. Biomarker Insights. 5, 9-20 (2010).
- Holmseth, S., et al. Specificity controls for immunocytochemistry: the antigen preadsorption test can lead to inaccurate assessment of antibody specificity. Journal of Histochemistry & Cytochemistry. 60 (3), 174-187 (2012).
- Burry, R. W. Controls for immunocytochemistry: an update. Journal of Histochemistry & Cytochemistry. 59 (1), 6-12 (2011).
- Hsu, S. M., Soban, E. Color modification of diaminobenzidine (DAB) precipitation by metallic ions and its application for double immunohistochemistry. Journal of Histochemistry & Cytochemistry. 30 (10), 1079-1082 (1982).
- Seth, A., Stemple, D. L., Barroso, I. The emerging use of zebrafish to model metabolic disease. Disease Models & Mechanisms. 6 (5), 1080-1088 (2013).
- Hsu, S. M., Raine, L., Fanger, H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. Journal of Histochemistry & Cytochemistry. 29 (4), 577-580 (1981).
- Cristino, L., et al. Obesity-driven synaptic remodeling affects endocannabinoid control of orexinergic neurons. Proceedings of the National Academy of Sciences of the United States of America. 110 (24), E2229-E2238 (2013).
- Imperatore, R., et al. Genetic deletion of monoacylglycerol lipase leads to impaired cannabinoid receptor CBR signaling and anxiety-like behavior. Journal of Neurochemistry. 135 (4), 799-813 (2015).
- Laperchia, C., et al. The excitatory/inhibitory input to orexin/hypocretin neuron soma undergoes day/night reorganization. Brain Structure and Function. 222 (8), 3847-3859 (2017).
- Eltoum, I., Fredenburgh, J., Grizzle, W. E. Advanced concepts in fixation: 1. Effects of fixation on immunohistochemistry, reversibility of fixation and recovery of proteins, nucleic acids, and other molecules from fixed and processed tissues. 2. Developmental methods of fixation. Journal of Histotechnology. 24 (3), 201-210 (2001).
- Mueller, C., et al. One-step preservation of phosphoproteins and tissue morphology at room temperature for diagnostic and research specimens. Public Library of Science One. 6 (8), e23780 (2011).
- Howat, W. J., Wilson, B. A. Tissue fixation and the effect of molecular fixatives on downstream staining procedures. Methods. 70 (1), 12-19 (2014).
- Dupre, M. P., Courtade-Saidi, M. Immunocytochemistry as an adjunct to diagnostic cytology. Annales de Pathologie. 32 (6), 433-437 (2012).
- Shi, S. R., Liu, C., Taylor, C. R. Standardization of Immunohistochemistry for Formalin-fixed, Paraffin-embedded Tissue Sections Based on the Antigen Retrieval Technique: From Experiments to Hypothesis. Journal of Histochemistry & Cytochemistry. 39, 741-748 (2006).
- Giorno, R. A comparison of two immunoperoxidase staining methods based on the avidin-biotin interaction. Diagnostic Immunology. 2 (3), 161-166 (1984).
- Ramos-Vara, J. A., Saeteele, J., Howard, G. C., Kaser, M. R. Immunohistochemistry. Making and Using Antibodies: A Practical Handbook. , 273-314 (2007).
- Jamur, M. C., Oliver, C. Permeabilization of cell membranes. Methods in Molecular Biology. 588, 63-66 (2010).
- Buchwalow, I., Somoloiva, V., Boecker, W., Tiemann, M. Nonspecific binding of antibodies in immunohistochemistry: fallacies and facts. Scientific Reports. 1, 28 (2011).
- Ansorg, A., Bornkessel, K., Witte, O. W., Urbach, A. Immunohistochemistry and multiple labeling with antibodies from the same host species to study adult hippocampal neurogenesis. Journal of Visualized Experiments. (98), (2015).
- Kalyuzhny, A. E. The dark side of the immunohistochemical moon: industry. Journal of Histochemistry & Cytochemistry. 57 (12), 1099-1101 (2009).
- Hewitt, S. M., Baskin, D. G., Frevert, C. W., Stahl, W. L., Rosa-Molinar, E. Controls for immunohistochemistry: the Histochemical Society’s standards of practice for validation of immunohistochemical assays. Endocrinology. 155 (3), 676-687 (2014).
- Ivell, R., Teerds, K., Hoffman, G. E. Proper application of antibodies for immunohistochemical detection: antibody crimes and how to prevent them. Journal of Histochemistry & Cytochemistry. 62 (10), 693-697 (2014).
- Stradleigh, T. W., Ishida, A. T. Fixation strategies for retinal immunohistochemistry. Progress in Retinal and Eye Research. 48, 181-202 (2015).
- Boi, G., Scalia, C. R., Gendusa, R., Ronchi, S., Cattoretti, G. Disaccharides Protect Antigens from Drying-Induced Damage in Routinely Processed Tissue Sections. Journal of Histochemistry & Cytochemistry. 64 (1), 18-31 (2016).
- Curran, R. C., Gregory, J. Demonstration of immunoglobulin in cryostat and paraffin sections of human tonsil by immunofluorescence and immunoperoxidase techniques. Effects of processing on immunohistochemical performance of tissues and on the use of proteolytic enzymes to unmask antigens in sections. Journal of Clinical Pathology. 31 (10), 974-983 (1978).
- O’Hurley, G., et al. Garbage in, garbage out: a critical evaluation of strategies used for validation of immunohistochemical biomarkers. Molecular Oncology. 8 (4), 783-798 (2014).
- Matos, L. L., Trufelli, D. C., de Matos, M. G., da Silva Pinhal, M. A. Immunohistochemistry as an important tool in biomarkers detection and clinical practice. Biomarker Insights. 5, 9-20 (2010).
- Mayersbach, H. V. Principles and limitations of immunohistochemical methods. Journal of Royal Microscopical Society. 87 (2), 295-308 (1967).
- Shi, S. R., Liu, C., Taylor, C. R. Standardization of immunohistochemistry for formalin-fixed, paraffin-embedded tissue sections based on the antigen-retrieval technique: from experiments to hypothesis. Journal of Histochemistry & Cytochemistry. 55 (2), 105-109 (2007).
- Dixon, A. R., et al. Recent developments in multiplexing techniques for immunohistochemistry. Expert Review of Molecular Diagnostics. 15 (9), 1171-1186 (2015).
- Mölne, J., Breimer, M. E., Svalander, C. T. Immunoperoxidase versus immunofluorescence in the assessment of human renal biopsies. American Journal of Kidney Diseases. 45 (4), 674-683 (2005).
- Gerdes, M. J., et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proceedings of the National Academy of Sciences of the United States of America. 110 (29), 11982-11987 (2013).
- Zhang, P., Lehmann, B. D., Shyr, Y., Guo, Y. The Utilization of Formalin Fixed-Paraffin-Embedded Specimens in High Throughput Genomic Studies. International Journal of Genomics. , 1926304 (2017).
- Xie, R., et al. Factors influencing the degradation of archival formalin-fixed paraffin-embedded tissue sections. Journal of Histochemistry & Cytochemistry. 59 (4), 356-365 (2011).
- Webster, J. D., Miller, M. A., Dusold, D., Ramos-Vara, J. Effects of prolonged formalin fixation on diagnostic immunohistochemistry in domestic animals. Journal of Histochemistry & Cytochemistry. 57 (8), 753-761 (2009).
- Drummen, G. P. Fluorescent probes and fluorescence (microscopy) techniques–illuminating biological and biomedical research. Molecules. 17 (12), 14067-14090 (2012).
- Marks, K. M., Nolan, G. P. Chemical labeling strategies for cell biology. Nature Methods. 3 (8), 591-596 (2006).
