All methods described here have been approved by the Institutional Animal Care and Use Committee (IACUC) of Touro University California (Protocol # TUCA003TE01X).
1. Obtaining X. laevis Embryos
- Obtain X. laevis embryos by natural mating of pairs of male and female adult frogs primed with human chorionic gonadotropin (HCG), by in vitro fertilization of eggs shed from female adult frogs primed with HCG, or by ordering directly (Table of Materials).
- Dejelly embryos obtained with a 2% cysteine solution at room temperature (Table of Materials)16.
- Collect 50−100 embryos in a large Petri dish. Remove the solution the embryos are in by decanting or using a plastic transfer pipette. Add 25 mL of 2% cysteine solution (0.5 g cysteine in 25 mL ddH2O, pH to 8.0) to the dish containing the embryos.
- Gently swirl the Petri dish containing the embryos in the cysteine solution until the jelly coats of the embryos fall off and the embryos collect in a clump in the center of the dish (5−10 min). At this point, slowly and gently pour off the cysteine solution into a waste beaker. Take care not to pour too many of the embryos in the waste beaker along with the cysteine solution.
- Rinse the embryos in the Petri dish 6x with 10% modified Mark’s Ringer solution (MMR) or other suitable solution (e.g., modified Barth’s Solution, MBS), swirling the dish each time the solution is replaced.
- Culture the embryos in 10% MMR until they reach developmental stages 22−2417. Xenopus embryos can be incubated at temperatures between 15−25 °C. The rate of development of the embryos depends on the temperature they are incubated at17.
NOTE: Xenopus embryos ordered from a catalogue usually arrive in the laboratory at developmental stages 20−24, so they can be dejellied and microinjected right away.
2. Preparing DNA Plasmids and Making a DNA/DOTAP Mixture
- Subclone DNA expression constructs into Xenopus expression vectors pCS2+ or pCS2+MT or derivatives thereof (originally constructed by D. Turner and R. Rupp)5,6,7. pCS2+ vectors contain a modified cytomegalovirus (CMV) promoter that facilitates gene expression in frogs.
- Amplify pCS2 plasmids containing GFP and/or genes of interest with miniprep kits (Table of Materials) following the standard procedure. In the final elution step of the miniprep protocol, perform a sequential elution of the DNA into ddH2O to yield a final concentration of >1 µg/µL.
- Store all pCS2 plasmids at -80 °C until ready to perform a microinjection/lipofection experiment, i.e., when embryos are at developmental stages 22−24.
- Thaw DNA plasmids to be lipofected at room temperature. Immediately prior to lipofection, briefly centrifuge DNA plasmids. This will prevent precipitate from forming in the DNA/DOTAP mixture that could clog the tip of the microcapillary pipette.
- Combine DNA plasmids with the DOTAP liposomal transfection reagent (Table of Materials) at a 1:3 (w/v) ratio9,10. For example, transfer 2 µg of DNA to a 1.5 mL microcentrifuge tube and add 6 µL of DOTAP, or transfer 3 µg of DNA in a microcentrifuge tube and add 9 µL of DOTAP.
- Once the DNA and DOTAP are combined, gently flick the microcentrifuge tube to mix the solution. The DNA/DOTAP solution should become slightly opaque after mixing.
- If two plasmids are to be lipofected together (e.g., pCS2-GFP with a second pCS2 plasmid containing a truncated or full-length version of a gene) in optic neurons, first combine the two plasmids (after briefly centrifuging both of them) at a 1:1 ratio, and then add DOTAP at a 1:3 (w/v) ratio. For example, combine 1 µg of pCS2-GFP with 1 µg of a second pCS2 plasmid and then add 6 µL of DOTAP.
NOTE: Studies have shown that lipofection of two plasmids into eyebuds of Xenopus embryos at these developmental stages will result in their co-expression in individual optic neurons9,10.
3. Loading a Microinjection Needle with DNA/DOTAP
- Gently clip the tip of a pulled glass microcapillary pipette with fine forceps (Table of Materials).
- Backfill the glass microcapillary pipette with mineral oil using a microfil such that a tiny drop of mineral oil appears at the clipped tip of the micropipette. Fill the microcapillary pipette halfway with mineral oil.
- Load the pulled glass microcapillary pipette that is now filled with mineral oil into a suitable injection holder connected to an injector. If using an injector (Table of Materials), eject the plunger halfway before loading the microcapillary pipette onto it. Once the microcapillary pipette is securely attached to the injector, extend the plunger to the full extent to confirm that the microcapillary pipette is strongly attached to the injector and does not move with the extension of the plunger.
- Transfer a 3 μL drop of the DNA/DOTAP mixture onto a cut square (1 inch square) sheet of paraffin paper.
- Under a stereo dissecting microscope, move the tip of the glass microcapillary pipette into the DNA/DOTAP drop.
- Slowly suck the DNA/DOTAP drop into the glass microcapillary pipette using the fill option on the injection apparatus. As the liquid is loaded into the microcapillary pipette, the drop will get smaller. Due to the slight opacity of the DNA/DOTAP solution, the boundary between the mineral oil and the DNA/DOTAP solution should be visible in the glass microcapillary pipette (Figure 1). If needed, stop filling the microcapillary pipette periodically to allow the pressure in the glass microcapillary pipette to recalibrate.
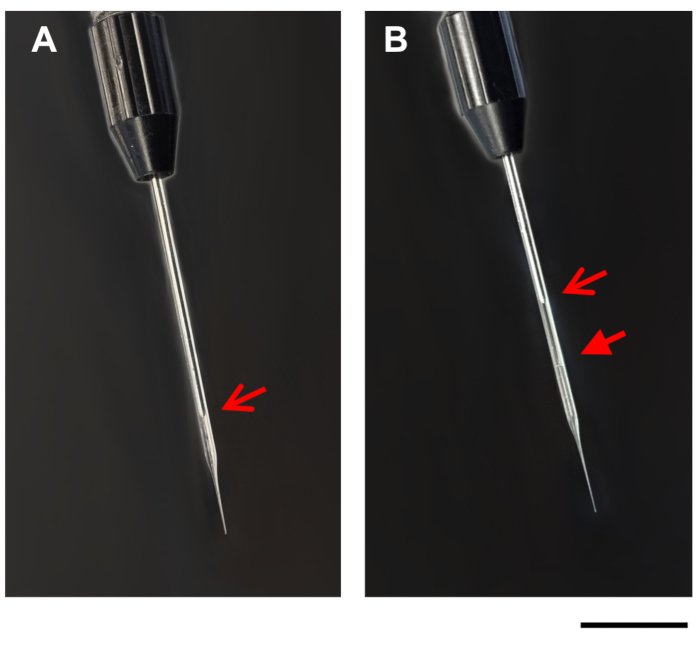
Figure 1: Images of microcapillary pipette. Images show a microcapillary pipette on the injection apparatus, before (A), and after (B) filling with DNA/DOTAP. Open arrows, tip of plunger in the microcapillary pipette (A,B). Closed arrow, line between mineral oil and DNA/DOTAP in the filled microcapillary pipette (B). Scale bar = 1 mm. Please click here to view a larger version of this figure.
4. Microinjecting DNA/DOTAP into Eyebuds of 1 Day Old Xenopus Embryos
- Manually devitellinize ten stage 20−24 Xenopus embryos with fine forceps in a 10 mm Petri dish filled with 0.1x MMR. Grasp the vitelline envelope at the waist to avoid injuring the embryos. With forceps in both the experimenter’s left and right hands, pop the bubble of the vitelline envelope and release the embryo from the vitelline envelope. Take care not to injure the embryos when removing the vitelline envelope.
NOTE: Beginning at stage 20, Xenopus embryos develop an indentation or ‘waist’ between the anterior and posterior halves of the embryo. This waist allows a gap to form between the vitelline envelope and the embryo at this position. - Use a plastic transfer pipette with a cut tip to transfer 5−10 devitellinized stage 22−24 embryos to a 10 mm Petri dish filled with 1x MMR.
NOTE: The higher salt solution in 1x MMR facilitates healing of puncture wounds that will result from the microinjection. - Under the stereomicroscope, grasp one of the devitellinized embryos in the Petri dish with forceps in each hand, and arrange the embryo so that its anterior pole is pointed up in the field of view. Orient the embryo so that it is lying laterally and one of its eyebuds (left or right) is facing upwards.
- Under the stereomicroscope, hold the embryo with the forceps in the experimenter’s non-dominant hand, and with the experimenter’s dominant hand introduce the tip of the glass micropipette into the eyebud (from the ventral or the dorsal side, depending on the half of the embryo that is currently being injected), just beneath the epidermis (Figure 2). Inject between 70−210 nL of the DNA/DOTAP solution. This can be done in several pulses, depending on the size of the pulse the injector is set to (usually 70 nL).
NOTE: The deepness of injection is very important for lipofection into optic neurons. If the position of the tip of the microcapillary is correctly inserted very superficially into the eyebud, then following microinjection, the grey epidermis overlying the eyebud will swell. If the position is too deep, the grey eyebud will not show any changes, and the frequency of expression of DNA in optic neurons will be lower. - Turn the embryo around and perform the same microinjection into the eyebud on the contralateral side of the embryo.
- Inject both eyebuds of 6−10 (or more) embryos in each experiment.
- After microinjection, store embryos in a Petri dish with 1x MMR for approximately 30 min to facilitate wound healing.
- After 30 min, transfer injected embryos into a 0.1x MMR solution with 0.001% bleaching agent (phenylthiocarbamide) to reduce pigmentation. Culture the embryos covered for approximately five days, until the embryos have developed into tadpoles at stages 46−4716.
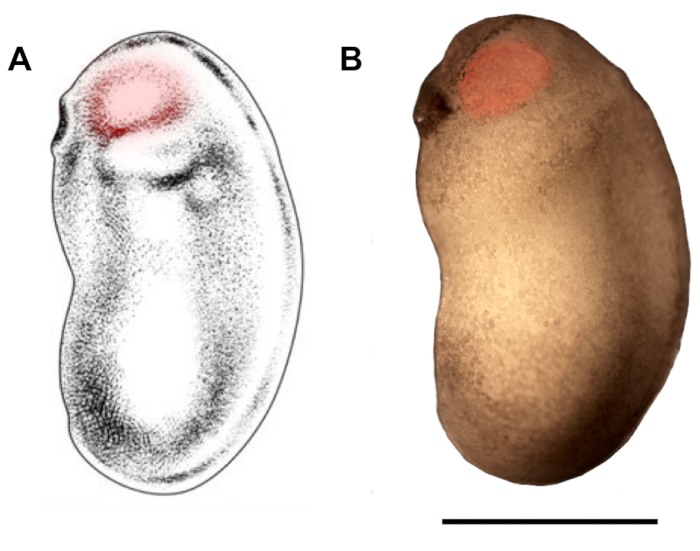
Figure 2: Demarcation of eyebud region for microinjection. Schematic (A) and photomicrograph (B) of X. laevis embryo at developmental stages 22/23 show eyebud region that should be targeted for microinjection (red highlights). Scale bar = 1 mm. Panel A has been modified from Zahn et al.18. Please click here to view a larger version of this figure.
5. Imaging of GFP Expressing Optic Axonal Arbors in Intact, Living Tadpoles
NOTE: When tadpoles that were lipofected with DNA reach developmental stages 46−47, they are ready for imaging.
- Prior to imaging, tadpoles must be anesthetized. To anesthetize tadpoles, transfer the lipofected tadpoles into a 0.02% tricaine solution in ddH2O in a 10 mm Petri dish. Wait 5−10 min until tadpoles become immobile. Verify that tadpoles are still alive by observing their beating hearts under a stereo dissecting microscope.
- Place one anesthetized tadpole into a custom-made silicone chamber on a glass slide and seal with a coverslip. The tadpole should tilt slightly so that one side (left or right) of its head is angled upwards and just barely touches the cover slip.
- Screen the half of the dorsal tectal midbrain of the tadpole that is tilted upwards at low magnification for GFP expressing optic axonal arbors.
NOTE: A widefield upright microscope equipped with an epilfuorescence illumination and an apochromatic objective lens (Table of Materials) can be used to screen for fluorescent arbors. - If the tectal hemisphere contains between one to three GFP expressing optic axonal arbors, capture a z-series of images of these arbors using a high contrast 40x air long working distance objective (Table of Materials). For each axonal arbor, capture 10−20 z-series slices at 1.5 µm intervals.
- In order to view axon arbors on the other side of the tectal midbrain, reload the tadpole in the silicon chamber so that it tilts to the other side and seal with a cover slip. Then repeat steps 5.3 and 5.4.
6. Reconstruction and Quantification of Optic Axonal Arbor Morphology
- Select an image stack that contains between one to three GFP expressing optic axonal arbors.
- Use the freehand drawing tool in graphic editing software (Table of Materials) to trace the portion of each optic axonal arbor visible in each z-slice. Tracing through the pieces of each arbor evident in each z-slice will create an accurate 2D projection of the arbor. Different colors can be used to trace distinct GFP expressing optic axonal arbors.
- Make all morphometric measurements on 2D reconstructions of the axonal arbors, with reference to the original z-series of images when needed7. Using Image J software (Table of Materials), measure morphological parameters such as number of branches (i.e., number of branch tips or branch points), total arbor branch length, length per branch, length and width of arbor, overall shape of arbor (L/W ratio, circularity), and angle of branches7.
The protocol described in this article yields a success rate of 30−60% of injected Xenopus embryos expressing GFP (alone or together with an additional DNA constructs) in one to ten optic axonal arbors. In Figure 3, we show representative confocal images of GFP expressing control and mutant optic axonal arbors in intact Xenopus tadpoles from our recently published study7. For this study, we cloned two domain mutants of APC (APCNTERM and APCβ-cat) into pCS2 plasmids, and co-injected these plasmids together with a pCS2-GFP labelling plasmid into eyebuds of one day old Xenopus embryos. Figure 4 shows results of several quantitative measurements we made on reconstructions of the control and APC mutant axonal arbors, including number of branches, total arbor branch length, and mean length of branches.
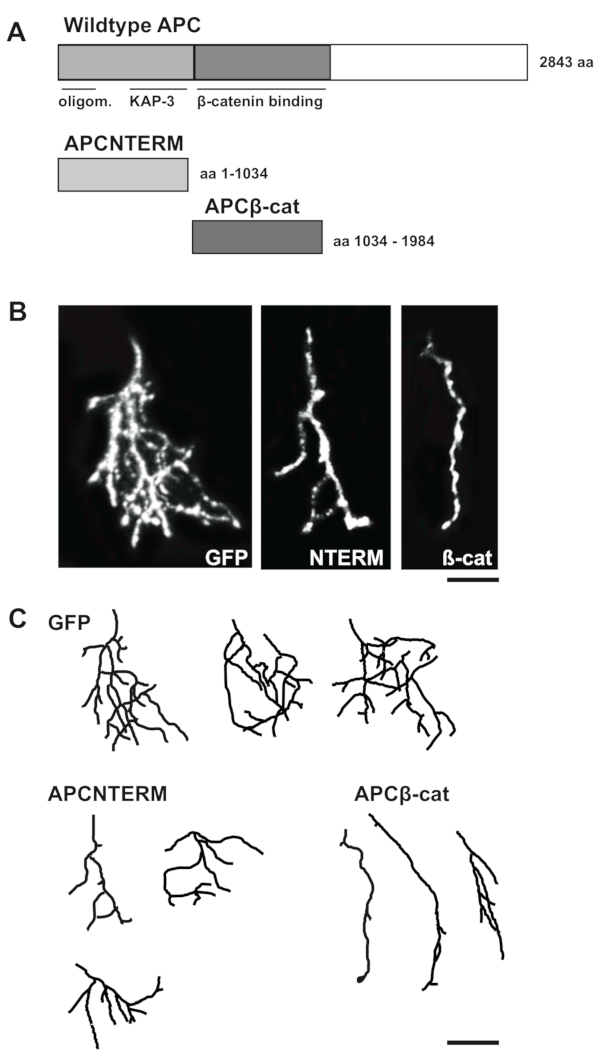
Figure 3: Representative images of GFP expressing control and mutant optic axonal arbors. (A) Schematic of mutants of APC N-terminal and central domain mutants that were cloned into pCS2 plasmids. (B) Representative confocal images of single GFP and GFP-APC mutant optic axonal arbors in tecta of intact, living Xenopus tadpoles. (C) Reconstructions of z-series images of GFP control and APC mutant optic axonal arbors. Scale bars = 30 µm (B), 40 µm (C). This figure has been modified from Jin et al.7. Please click here to view a larger version of this figure.
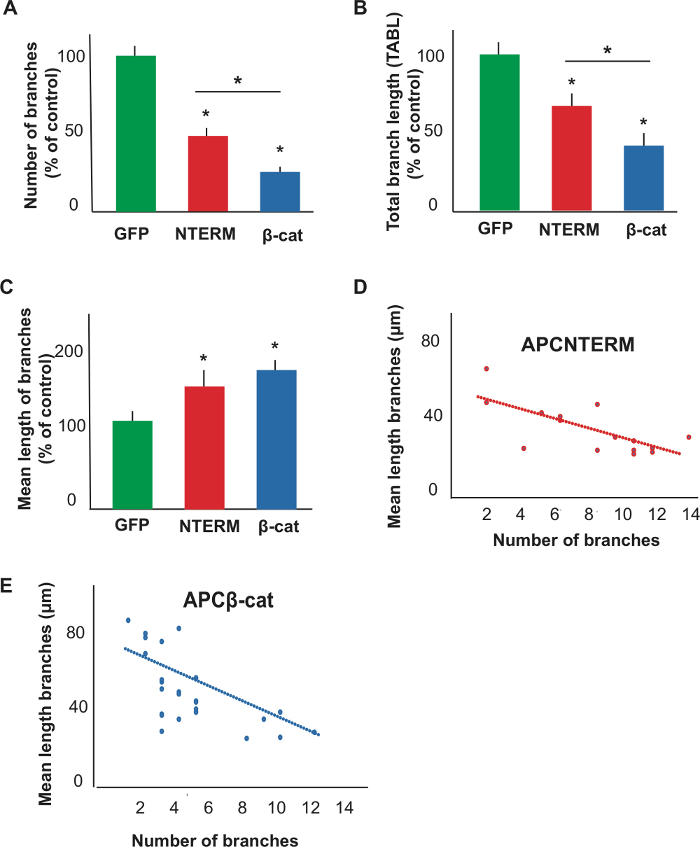
Figure 4: Quantification of morphologies of reconstructions of control and mutant axonal arbors. Plots of number of branches (A), total arbor branch length (B), and mean branch length (C) confirm observed differences between control and APC mutant expressing axonal arbors. Data in panels A−C is shown as percent of control mean with SEM. *above data bar or line indicates p < 0.05. Additional scatter plots of number of branches versus mean branch length with regression lines show inverse correlation between these parameters in optic axonal arbors expressing APC domains (D, E). Sample numbers: (A) GFP-12, APCNTERM–18 APCβ-cat-25; (B) GFP-12, APCNTERM-16, APCβ-cat-25; (C) GFP-11, APCNTERM-16, APCβ-cat-25. This figure has been modified from Jin et al.7. Please click here to view a larger version of this figure.
