Descrevemos uma abordagem de imagem total de reflexão interna fluorescência-número e brilho (TIRF-N&B) para determinar o estado oligomérico médio das moléculas receptoras na membrana plasmática das células vivas, com o objetivo de vincular a montagem do receptor dinâmica para a função biológica das proteínas (Figura 1).
Após a ligação extracelular do ligand, os receptores iniciam a transdução de sinal intracelular dependendo de sua conformação, oligomerização, potenciais co-receptores e composição da membrana. Apesar da importância e ubiquidade da oligomerização do receptor, reconhecida como um evento-chave na sinalização celular1,2,3,4,5,6, 7,poucos métodos podem detectar eventos de agrupamento e medir o grau de agrupamento experimentalmente8,9. O volume confocal (x, y – 300 nm, z 900 nm) é insuficientemente resolvido para provar a interação molecular e stoichiometria, mesmo após a otimização por algoritmos de restauração de imagem10. A composição sub-unitária de oligomeros proteicos não pode ser resolvida de forma puramente espacial, mesmo por métodos de super-resolução em x,y resolução de 20-70 nm, como PALM11, STORM12,e STED13. Além disso, sua resolução temporal (na ordem dos minutos por imagem) não pode seguir a cinética na faixa de segundos. Único molécula passo-branqueamento resolve a stoichiometria de oligomeros de proteína apenas se eles são imóvel14.
Um dos métodos mais versáteis para medir a densidade e o oligomerização de proteínas fluorescentes marcadas dentro de imagens individuais é a análise de distribuição de intensidade espacial (SpIDA), que depende da amostragem espacial. É aplicável a células quimicamente fixas e vivas, e permite a análise de várias regiões de interesse da célula simultaneamente usando microscopia fluorescência padrão15. Alternativamente, métodos momentâneos, como espectroscopia fluorescência-correlação (FCS)16,fótons contando histograma (PCH)17, e Número e Brilho (N&B)18,19, são adequados para oligomer quantitativo Medidas. Esses métodos analisam as flutuações de intensidade da fluorescência que podem ser observadas no tempo em que os fluorofforas se difundem dentro e fora de um volume de iluminação. As amplitudes das flutuações de intensidade podem ser descritas exclusivamente pelo brilho molecular do fluorofforre (ε) e pelo número médio de fluorofônicos (n) dentro do volume de iluminação17 (Figura 2). Normalmente, o coeficiente de difusão dos fluorofforos e o número médio de moléculas (inversamente relacionadas ao valor G(0) dentro do volume de iluminação podem ser obtidos pela FCS20. No entanto, uma vez que o tempo de difusão apenas escalas com a raiz cúbica da massa, FCS não é suficientemente sensível para detectar alterações na massa molecular21. Na prática, a FCS de cor única não consegue detectar a dimerização dos receptores de membrana. PCH resolve misturas de diferentes oligomeros com precisão. Usando mais de dois momentos da distribuição da amplitude, detecta moléculas do brilho diferente que ocupam o mesmo volume da iluminação. Digitalização FCS22 e desenvolvimentos, como a interessante par-correlação de brilho molecular (pCOMB) abordagem23, introduzido para estender a gama de aplicabilidade dos métodos de correlação fluorescência em sistemas biológicos24 , permanecem métodos de ponto único sem a capacidade de medições rápidas em uma grande área de uma célula, exigindo muitas observações consecutivas em cada pixel e aquisição de dados na ordem de segundos.
A N&B é uma versão simplificada do PCH que considera apenas o primeiro e segundo momentos da amplitude da distribuição de fluorescência, ou seja, a intensidade média, e a variação, σ2 (Figura 2)18,19 e, por causa disso, não pode determinar a fração molar de oligomeros desconhecidos em uma mistura, mas estima somente o estado médio do oligomerization da mistura. No entanto, a N&B tem a vantagem de trabalhar com uma série de imagens de imagens de células vivas relativamente menores do que a PCH em uma base pixel-a-pixel, simplesmente monitorando as flutuações no tempo da intensidade da fluorescência. Como a N&B reduz o tempo por pixel para alguns microssegundos, ela pode seguir cinética de oligomerização rápida sobre grandes áreas celulares, permitindo a aquisição de imagem em uma escala de tempo de segundos em microscopia de varredura de raster (por exemplo, confocal, 2 fótons) e milissegundos em microscopia baseada em câmera (por exemplo, TIRFM).
Vários relatos demonstraram a capacidade da N&B de quantificar o número de subunidades em aglomerados de proteínas por meio de imagens de regiões celulares estendidas. Os clusters Paxillin-EGFP foram detectados nos locais de adesão nas células CHO-K125,e a agregação intracelular do peptídeo httex1p patogênico foi descrita nas células COS-726. A N&B foi aplicada para seguir a oligomerização ativa da ligand do receptorErbB 27,e o efeito do ligand FGF21 em Klothob (KLB) e fgfr1c em célulasHeLa 28. A combinação de imagens tirf e análise de N&B foi usada para mostrar que a dinamina-2 é principalmente tetramérica em toda a membrana celular29. Aplicamos a N&B a imagens de varredura de raster e TIRF para provar a dimerização conduzida por ligand dos receptores de membrana celular uPAR e FGFR130,31.
Métodos de correlação de fluorescência, como N&B, FCS e PCH, são baseados na noção de que, em um volume aberto, o número de bolhas de partículas segue uma distribuição de Poisson. Como somente os fótons que os fluorofônicos emitem podem ser detectados, o valor médio para uma intensidade  de fluorescência medida versus o tempo em um pixel da imagem, é o produto do número médio de fluorofosforas no volume de iluminação, n, e sua brilho molecular, ε17:
de fluorescência medida versus o tempo em um pixel da imagem, é o produto do número médio de fluorofosforas no volume de iluminação, n, e sua brilho molecular, ε17:

onde o ε é expresso como o número de fótons emitidos por unidade de tempo (convencionalmente por segundo) por molécula quando a molécula está no centro do volume de iluminação.
O brilho é propriedade de cada fluorofofóbico em uma determinada aquisição criada, enquanto a intensidade é a soma de todas as contribuições de todos os fluorofos. Em competições biológicas, o brilho aumentará com o aumento do número de fluorofos que flutuam junto, dando a informação no estado do oligomerization da proteína fluorescente-etiquetada. As amplitudes de flutuação em um determinado pixel é medido a partir da variação do sinal de fluorescência, σ2:

Onde a média do quadrado  de intensidade, e o quadrado
de intensidade, e o quadrado  da média de intensidade, são computados a partir dos valores de intensidade individual em cada pixel de cada quadro:
da média de intensidade, são computados a partir dos valores de intensidade individual em cada pixel de cada quadro:
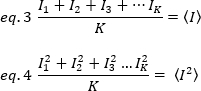
onde K é o número de quadros totais na série de tempo. Experimentalmente, é necessário calcular para toda a série de imagens a variação que descreve a dispersão dos valores de intensidade individual em cada pixel de uma única imagem em torno do valor de intensidade média. A variação inclui todas as flutuações de diferentes origens. Em uma primeira aproximação, a variação devido às partículas de difusão no volume de iluminação, σ20,pode ser separada da variância devido ao ruído do tiro do detector, σ2d. As duas variações são independentes; assim, a variação total é dada por sua soma:

A variação, devido a flutuações moleculares dentro e fora do volume de detecção, é linearmente dependente do brilho e intensidade moleculares:

Reorganizando eq. 6 de acordo com o eq. 1:

De acordo com o conceito típico de espectroscopia de correlação de fluorescência, a equação 7 afirma que a variação devido ao número de flutuações depende do quadrado do brilho das partículas.
Em seguida, a variação devido às flutuações do detector é uma função linear da intensidade detectada, a suposição de que o detector é operado abaixo de seu limite de saturação19:

No caso dos detectores de contagem de fótons =1e c=0, assim a variação do detector é igual à intensidade média:

Para aplicar esses conceitos a medições reais em células vivas, Gratton e colegas18 definem o brilho aparente, B, para cada pixel como a proporção da variação sobre a intensidade média:

B é o parâmetro que é medido experimentalmente. Neste trabalho, imagens de séries de tempo de receptores FGFR1 na membrana plasmática das células HeLa são capturadas pela microscopia TIRF e o brilho aparente médio, B, é determinado pela análise de N&B. Então, após a adição de FGF2, séries de tempo consecutivos são capturadas para acompanhar as mudanças na auto-montagem das moléculas receptoras na superfície da membrana após a estimulação do receptor com o ligand canônico.
No entanto, uma vez que o detector do microscópio TIRF é uma câmera EMCCD, a expressão para o brilho aparente precisa ser modificada como19:

onde o deslocamento é o deslocamento da intensidade da eletrônica da deteção que é uma característica das ajustes do detetor. A variação e a intensidade média de um detector analógico são, respectivamente, dadas por:
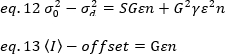
onde G é o ganho analógico em níveis digitais (DL/fótons), S, os níveis digitais por fóton19, é dado pela inclinação de uma intensidade versus variação parcela para uma fonte de luz com intensidade constante (sem flutuações temporais). O fator γ está relacionado à forma do volume de detecção de pixels. De acordo com Hassler et al.32,o fator γ é igual a 0,3 para imagens TIRF trabalhando no ganho máximo da câmera de detecção19. Os parâmetros de deslocamento, S e G são características da câmera e do microscópio. O brilho aparente, B, é obtido reorganizando eq. 11 de acordo com eq. 12 e 13:

Experimentalmente, o ε é uma função complexa da intensidade do laser e da eficiência de detecção do sistema. No entanto, uma vez que B / S é linearmente dependente de ε, só é importante determinar o valor relativo do ε para um determinado modo de detecção:

onde o ε’ é proporcional ao ε. Ainda assim, uma calibração é realizada usando uma referência interna.
