Quelle: Tilde Andersson1, Rolf Lood1
1 Institut für Klinische Wissenschaften Lund, Abteilung für Infektionsmedizin, Biomedical Center, Universität Lund, 221 00 Lund, Schweden
Viren, die prokaryotische Organismen infizieren, bakteriopische oder einfach Phagen genannt, wurden Anfang des20. Jahrhunderts durch Twort (1) und d’Hérelle (2) unabhängig voneinander identifiziert. Phages sind seitdem weithin anerkannt für ihren therapeutischen Wert (3) und ihren Einfluss auf den Menschen (4) sowie globale Ökosysteme (5). Die derzeitigen Bedenken haben ein erneutes Interesse an der Verwendung von Phagen als Alternative zu modernen Antibiotika bei der Behandlung von Infektionskrankheiten geschürt (6). Im Wesentlichen beruht die gesamte Phagenforschung auf der Fähigkeit, Viren zu reinigen und zu quantifizieren, auch als viraler Titer bekannt. Ursprünglich 1952 beschrieben, war dies der Zweck des Plaque-Assays (7). Jahrzehnte und mehrere technologische Fortschritte später, bleibt der Plaque-Assay eine der zuverlässigsten Methoden zur Bestimmung von viralem Titer (8).
Bakteriophagen erbestehen, indem sie ihr genetisches Material in Wirtszellen injizieren, die Maschinen für die Produktion neuer Phagenpartikel entführen und schließlich dazu führen, dass der Wirt zahlreiche Nachkommenvirionen durch Zelllyse freisetzt. Aufgrund ihrer winzigen Größe können Bakteriophagen nicht allein mit der Lichtmikroskopie beobachtet werden; Daher ist eine Rasterelektronenmikroskopie erforderlich (Abbildung 1). Darüber hinaus können Phagen nicht auf Ernährungs-Agar-Platten wie Bakterien kultiviert werden, da sie Wirtszellen benötigen, um Beute zu machen.
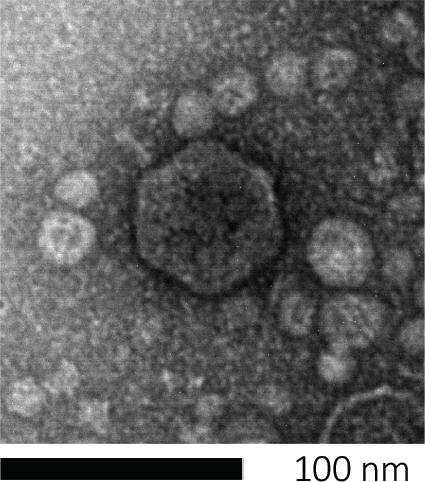
Abbildung 1: Die Morphologie eines Bakteriophagens, hier am Beispiel eines E. coli-Phagens, kann mit Derkelektronenmikroskopie untersucht werden. Die meisten Bakteriophagen gehören zu Caudoviralen (tailed bacteriophages). Dieser besondere Phagen hat eine sehr kurze Schwanzstruktur und einen Ikosaederkopf, der ihn in die Familie der Podoviren eingibt.
Der Plaque-Assay (Abbildung 2) basiert auf der Einbindung von Wirtszellen, bevorzugt im Logphasenwachstum, in das Medium. Dies schafft eine dichte, trübe Schicht von Bakterien in der Lage, virales Wachstum zu erhalten. Ein isolierter Phagen kann anschließend eine Zelle infizieren, replizieren und lysieren. Mit jeder lysierten Zelle werden mehrere benachbarte Zellen sofort infiziert. Mehrere Zyklen in, eine klare Zone (eine Plaque) kann in der ansonsten trüben Platte beobachtet werden (Abbildung 2B/Abbildung 3A), was auf das Vorhandensein eines anfangs einzelnen Bakteriophagenpartikels hinweist. Die Anzahl der Plaqueformeinheiten pro Volumen(d.h. PFU/ml) einer Probe kann somit anhand der Anzahl der erzeugten Plaques bestimmt werden.
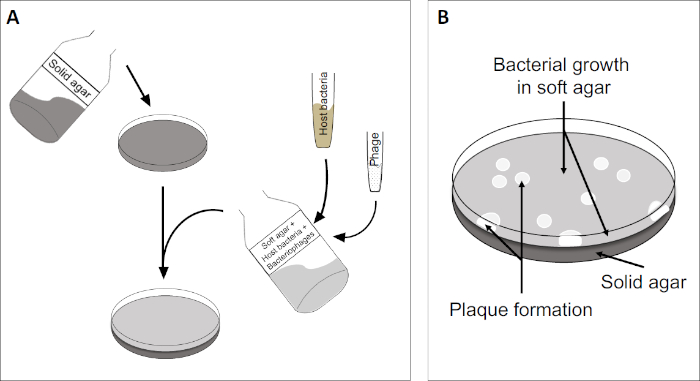
Abbildung 2: Tests auf Plaqueforming Units (PFU) ist eine gängige Methode zur Bestimmung der Anzahl der Bakteriophagen in einer Probe. (A) Die Basis einer sterilen Petrischale ist mit einem geeigneten festen Nährmedium bedeckt, gefolgt von einer Mischung aus weichen Medien, anfälligen Wirtszellen und einer Verdünnung der ursprünglichen Bakteriophagenprobe. Beachten Sie, dass die Phagensuspension in einigen Fällen auch gleichmäßig über die Oberfläche des bereits erstarrten weichen Agars verteilt werden könnte. (B) Das Wachstum der Wirtsbakterien bildet einen Rasen von Zellen in der oberen Agarschicht. Die Bakteriophagenreplikation erzeugt klare Zonen oder Plaques, die durch die Leierzelllyse verursacht werden.
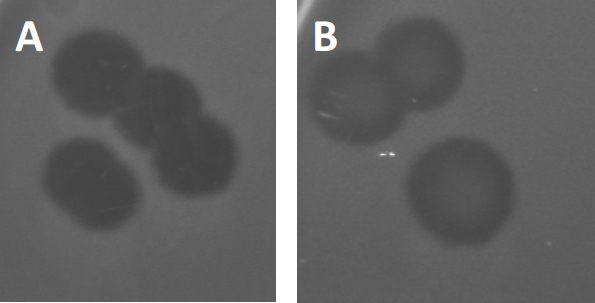
Abbildung 3: Die Ergebnisse der PFU-Tests zeigen mehrere Plaques, die durch Bakteriophagen erzeugt werden. Aufgrund der Lyse empfänglicher Wirtszellen können Plaques als Clearingzonen im bakteriellen Rasen angesehen werden, entweder mit (A) voller Clearance oder (B) teilweisem Nachwachsen, das durch die Erzeugung resistenter Bakterien (oder möglicherweise durch gemäßigte Phagen in der lysogene Zyklus).
Bestimmte gemäßigte Phagen können zusätzlich zu dem früher beschriebenen lytischen Wachstum einen sogenannten lysogenen Lebenszyklus annehmen. In der Lysogenesie nimmt das Virus einen latenten Zustand durch DieInintegration seines genetischen Materials in das Genom der Wirtszelle (9) an, was häufig Resistenzen gegen weitere Phageninfektionen verleiht. Dies zeigt sich manchmal durch eine leichte Trübung der Plaque (Abbildung 3B). Es ist jedoch erwähnenswert, dass Plaques auch durch das Wiederwachstum von Bakterien, die Resistenz gegen den Phagen unabhängig von früheren Phageninfektionen entwickelt haben, verschwommen erscheinen können.
Viren können nur an eine begrenzte Anzahl von Wirtsbakterien anhängen oder adsorbieren (10). Die Wirtsbereiche werden durch intrazelluläre antivirale Strategien wie das CRISPR-Cas-System (11) weiter begrenzt. Die Resistenz/Empfindlichkeit gegenüber bestimmten Phagen, die von bakteriellen Untergruppen angezeigt werden, wurde in der Vergangenheit verwendet, um Bakterienstämme in verschiedene Phagentypen zu kategorisieren (Abbildung 4). Obwohl die Wirksamkeit dieser Methode nun durch neuartige Sequenzierungstechniken übertroffen wurde, kann die Phagentypisierung dennoch wertvolle Informationen über Bakterien-Phagen-Wechselwirkungen liefern, z. B. was die Entwicklung eines Phagencocktails für den klinischen Einsatz erleichtert. .
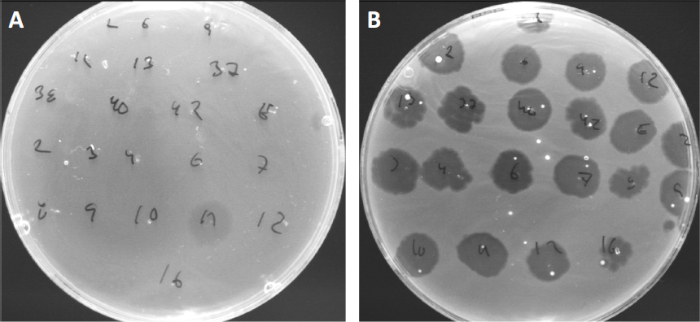
Abbildung 4: Phage-Empfindlichkeit verschiedener Bakterienstämme. Weiche Agarplatten mit Cutibacterium acnes Stamm (A) AD27 und (B) AD35, wurden mit 21 verschiedenen C. acnes Bakteriophagen gesichtet. Nur Phagen 11 waren in der Lage, AD27 zu infizieren und zu töten, während Stamm AD35 Empfindlichkeit gegenüber allen Phagen zeigte. Diese Technik, die als Phagentypisierung bezeichnet wird, kann verwendet werden, um Bakterienarten und Stämme in verschiedene Untergruppen zu unterteilen, basierend auf der Phagenanfälligkeit.