1. Method 1: Producing and purifying recombinant HAT1/Rbap46 complex
- Thawing, recovering, and expanding HEK293f cells
- Thaw 1-10 million HEK293f mammalian cells26 into 30 mL freestyle 293 expression media in a 100 mL flask. Incubate in 8% CO2 at 37 °C while rotating at 60 rpm.
- The next day, count and check cell viability, then adjust rotation speed to 120 rpm. Expand to 300 mL culture in a 1 L flask, maintaining seeding density at 500,000 cells/mL and splitting cells before density exceeds 3 x 106 cells/mL.
- HEK293f transfection
- Seed 5 x 105 cells/mL in 300 mL culture in a 1 L flask and culture for 24 h. The next day, count cells to ensure they are between 7.5 x 105 to 1.2 x 106 cells/mL.
- Prepare a mixture of 300 µg of pHEK-FLAG-HAT1 and 300 µg of pHEK-Rbap46 plasmid DNAs in 30 mL of PBS. Add 1.2 mL of polyethylenimine (PEI) to the DNA/PBS and mix. Incubate for 20 min at RT. Add the DNA/PBS/PEI mix to the 300 mL culture and incubate at 37 °C for 48 h at 120 rpm.
- Harvesting cells
- Count cells to ensure they fall between 1.7 x 106 to 2.5 x 106 cells/mL. At this cell density, add forskolin to a final concentration of 12.5 µM to activate HAT1 and incubate cells for 30 min, 37 °C, 120 rpm.
- Pellet the cells at 300 x g for 5 min, wash once with 30 mL of PBS, and snap-freeze in liquid N2. Store at -80 °C until protein purification or proceed directly to purification.
- HAT1/Rbap46 complex FLAG-bead purification
- Lyse cells and prepare protein extract.
- Prepare lysis buffer by adding one tablet of protease inhibitor cocktail (PIC) to 50 mL of RSB-500 (20 mM Tris-HCl pH 7.5, 500 mM NaCl, 25 mM MgCl2).
- Thaw the cell pellet from 300 mL culture on ice and lyse with 40 mL of ice-cold lysis buffer with 0.1% Triton X-100. Sonicate on ice, then spin down at 10,000 x g for 10 min at 4°C. Collect all supernatant into one 50 mL tube on ice, which now is the protein extract.
- Prepare FLAG beads.
- Split the FLAG beads into four 15 mL conical tubes by adding 400 µL of M2-FLAG agarose bead slurry into each tube containing 5 mL of 0.1 M Glycine pH 3.5, 0.01% Triton X-100. Shake each tube vigorously for 5 s by hand.
- Pulse centrifuge for 30 s at 1000 x g at 4 °C, aspirate supernatant, wash twice with 5 mL of RSB-500 + 0.1% Triton X-100 (Tx-100; no protease inhibitor), and incubate on ice.
- Perform FLAG immunoprecipitation.
- Add 10 mL of protein extract to each 15 mL conical tube containing washed beads. Incubate the tubes at 4 °C with inverted rotation for at least 90 min or overnight.
- Wash the bound beads 5x in 10 mL of RSB-500 + 0.1% Tx-100. For each wash, invert the tube 2x-5x to resuspend beads, then pellet at 1000 x g for 1 min at 4 °C. Finally, wash 1x in RSB-100 (20 mM Tris-HCl pH 7.5, 100 mM NaCl, 25 mM MgCl2) + 0.1% Tx-100.
- FLAG peptide elution: Elute the HAT1 complex in 1.5 mL of Elution Buffer (EB) containing 0.5 mg/mL FLAG peptide. Incubate at 4 °C for at least 1 h or overnight. Pulse centrifuge the eluted beads at 1000 x g for 30 s at 4 °C and collect the supernatant, which contains the purified HAT1/Rbap46 complex.
- Concentrate protein and remove FLAG peptide.
- Add eluate to a 20 mL, 10,000 Dalton cutoff filter tube. Bring the volume up to 15 mL with EB. Spin at 2500 x g for 15 min at 4 °C, repeating twice with an additional 15 mL of EB each time.
- Recover the final eluate from the filter tube and check the protein concentration by A260. Expect a concentration of 1 mg total protein in 6 mL of EB (per 300 mL starting culture). Check protein purity by SDS-PAGE, Coomassie, and immunoblotting, and store aliquots at -80 °C.
- Lyse cells and prepare protein extract.
2. Method 2: HAT1 acetyl-click standard curve
- Preparing standard curve
- Synthesize a positive control H4 N-terminal peptide with the sequence: SCRG[Pra]GGKGLG[Pra]GGAKRHRKVLRGG[Lys(Biotin)], where [Pra] denotes Propargylglycine.
- Resuspend the Pra peptide to 0.1 mg/mL in DMSO. Mix the Pra peptide with biotinylated histone H4 peptide (1-23-GGK-biotin) to create a standard curve (Figure 2; volumes found in Table 1). Standard curves may be made fresh prior to plate binding or, if made in advance, mixed with 20 µL of 8 M urea and stored at -20 °C until step 3.3.
3. Method 3: HAT1 acetyl-click assay
- Assembling acetylation reactions with test inhibitors
- Assemble acetylation reactions in duplicate in 0.2 mL PCR tubes or 96-well PCR plates from the following components: biotinylated histone H4 peptide (1-23-GGK-biotin) resuspended in DMSO to 0.1 mg/mL (34.8 µM), HAT1 enzyme pre-diluted in EB, 20x buffer (1M Tris pH 8.5, 0.1% NP40), 2 mM DTT, 4-pentynoyl-CoA dissolved in water to 1 mg/mL (1 mM). A 20 µL reaction comprises 10 µL of enzyme, 1 µL of H4 peptide, 1 µL of 20x buffer, 1 µL of DTT pre-mixed and aliquoted to wells of a 96-well PCR plate on ice.
- Add 1 µL of DMSO (negative control) or test compound dissolved at 1-10 µM, or H4K12CoA25 (or suitable positive control inhibitor), per well. Mix by gentle pipetting and incubate for 10 min on ice to allow enzyme: inhibitor complexes to form.
- Acetylation reaction continuation
- Combine 2 µL of 4-pentynoyl-CoA with 4 µL of water, then add to the wells. Gently mix by pipetting, centrifuge at 300 x g for 30 s at RT to collect the contents, and incubate at 37 °C for 1 h in capped tubes or plates with resealable foil.
- Process the contents directly for reaction products or quench with 20 µL of 8 M urea and store at -20 °C until processing.
- Peptide binding to Neutravidin plate
- Add reaction contents with or without urea to bovine serum albumin (BSA) pre-blocked black Neutravidin-coated 96-well plates containing 80 µL of PBST (PBS + 0.1% Tween-20) per well.
- Add standard curve peptides in duplicate to their own wells containing 80 µL of PBST. Bind peptides with gentle orbital shaking for 1 h at room temperature (RT).
- Wash: After binding is complete, aspirate the liquid from the wells and wash the wells with 200 µL of PBST 3x 15 strokes (180 µL stroke volume) using a plate washer.
- Click chemistry reaction
- Prepare the reagents for the click reaction as follows: 100 mM Tris(3-hydroxypropyltriazolymethyl)amine (THPTA) ligand in water and 20 mM CuSO4 in water. THPTA prevents Cu(II) catalyzed hydrolysis and quenches radicals and peroxides generated from O2/Cu/ascorbate.
- Add the THPTA/Cu mixture to 300 mM sodium ascorbate in water and 2.5 mM biotin azide in DMSO. One-click reaction contains 140 µL of PBS, 10 µL of THPTA, 10 µL of CuSO4, 10 µL of sodium ascorbate, 20 µL of biotin-azide. Always mix the click reagents fresh, then dispense 190 µL to each well, seal the plate, and incubate at 37°C for 1 h.
- Wash: Aspirate the liquid from the wells and wash the plate 3x with PBST (180 µL stroke volume, 15 strokes for each wash).
- Streptavidin-horseradish peroxidase binding: Dilute streptavidin-HRP (0.224 mg/mL) 1:10 in streptavidin (0.224 mg/mL), then further dilute 1:1000 in PBST. Add Steptavidin-HRP: streptavidin mix (100 µL per well) and incubate at RT for 1 h with gentle orbital shaking.
- Wash 3x with PBST as in the previous steps.
- Amplex red oxidation
- Combine the Amplex red detection reagents as follows: 4.45 mL of NaHPO4 buffer (1x = 50 mM final concentration), 50 µL amplex red (20 mM diluted in DMSO), 500 µL of diluted H2O2.
- Dilute H2O2 from 30% stock to 3% in 1x NaHPO4 buffer, then add 22.7 µL of 3% H2O2 into 977 µL of 1x NaHPO4 buffer (this is the H2O2 used for the amplex reaction). Add 100 µL of the amplex red reaction mixture per well, incubate at RT for 30 min protected from light, then detect the fluorescence excitation/emission 571/585 nm using a standard fluorescence plate reader.
- Calculation of enzyme inhibition: Calculate percent inhibition (Figure 3) according to the following formula:
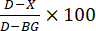 , where D is the fluorescence value of control reactions treated with DMSO only, X is the value of reactions treated with test compounds, and BG is the value of background wells (H4 peptide control, no enzyme added).
, where D is the fluorescence value of control reactions treated with DMSO only, X is the value of reactions treated with test compounds, and BG is the value of background wells (H4 peptide control, no enzyme added). - Dose curve: Select test compounds that are found to inhibit HAT1 enzyme activity for repeat assays, with the compound serially diluted. Determine the compound dilutions empirically. For example, start with 2 mM stock concentration, then serially dilute 1:3 for eight dilutions. Then dilute 1:20 into enzyme assays yielding 100 µM top dose.
- Plotting the curve: Use least squares regression to fit dose-response inhibition curves (using percent inhibition values from step 3.10) in data analysis software (GraphPad Prism) and derive IC50 values for each compound (Figure 4).
- Acetylation reaction with acetyl-CoA:
- Use this to confirm enzyme activity with the native co-factor acetyl-CoA instead of 4-pentynoyl-CoA. Carry out HAT1 acetylation assays as described in steps 3.1-3.2 with acetyl-CoA in place of 4-pentynoyl-CoA.
- Spot the reaction products onto nitrocellulose membranes (1-2 µL), allow them to dry, then dot-blot with anti-H4-lysine-12-acetyl or anti-H4-lysine-5-acetyl antibodies. Quantify the immunoblot signal by densitometry.
Standard curves in duplicate (16 wells) should be included on every plate to ensure proper assay performance. Standard curve data should be set up in table form, with a range of 100% to 0% according to the ratio of Pra-containing peptide to native H4 peptide in solution (Table 1). Amplex red signal will be the highest in 100% pra/0% native H4 peptide wells, and lowest in 0% pra/100% native H4 peptide wells. After fluorescence has been detected and wells are averaged, the resulting standards graph should fit a straight line (Figure 2), although saturation of signal at concentrations above 50% Pra can be observed. Improper mixing of Pra to H4 control peptides may result in little to no amplex red signal or nonlinear changes in amplex red signal between 0-50% Pra. Proper signal and curve results indicate that the click chemistry reaction has occurred, and the data can be analyzed accordingly.
It is important to always include triplicate control reactions: DMSO-treated reactions serve as negative controls, and H4K12CoA-treated reactions serve as positive controls for enzyme inhibition, respectively. These should be included on every plate to ensure robust assay performance and allow for Percent Inhibition calculations. Positive control wells that contain 1 µM of H4K12CoA should have a low amplex red signal, which is similar to the 0% Pra/100% H4 peptide wells. Negative control wells containing DMSO should have a signal that is similar to the fluorescent output of wells with 50% Pra/50% H4 peptide. Test compound wells will contain varying amplex signals. Average duplicate, or triplicate, wells to get a mean value of the signal with standard error.
Calculate enzyme inhibition with the formula above where D is the DMSO fluorescent output, X is the experimental output, and BG is the output from the H4 peptide without enzyme or the 0% well on the standard curve. Fluorescence output from the test compound reactions will be less than DMSO if the test compound inhibits enzyme activity and will result in positive inhibition percent values. The opposite effect will be seen in reactions where no enzyme inhibition has occurred. Example data of test compounds screened at two concentrations (1 µM and 10 µM) are depicted in Figure 3. Serial dilutions of the compounds can also be tested using this assay, where the IC50 of the compound can be found with non-linear curve fitting of the data (Figure 4).
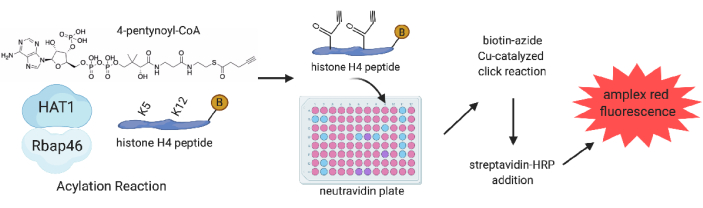
Figure 1: HAT1 acetylation/acylation click chemistry schematic. The HAT1 Acetylation click reaction occurs with purified HAT1 and Rbap46 protein in a solution with biotinylated histone H4 peptide, 4-pentynoyl-CoA, and test compounds. After reactions are completed, the solution is bound to a neutravidin-coated plate, and click chemistry with copper sulfate and biotin-azide is completed. The final addition of streptavidin-HRP and amplex red results in red fluorescent output. Comparison of amplex red fluorescence intensities determines HAT1 acetylation activity. This figure has been modified with permission from Gaddameedi et al.24. Please click here to view a larger version of this figure.
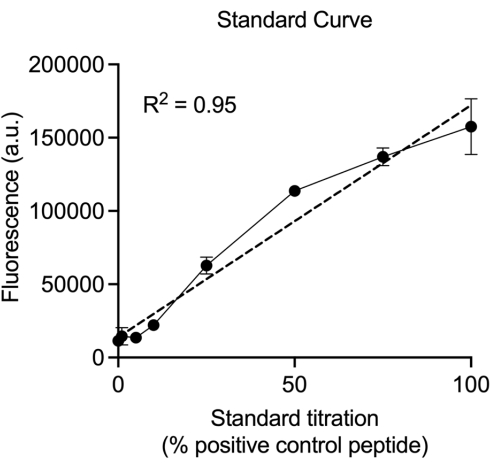
Figure 2: Example standard curve. After averaging the standard curve values for each pair, graph the means ± SEM against the positive control Pra peptide percentages. The resulting curve should resemble the shown curve and fit a linear line. This assay has a limit of detection (LOD) of 1.8% ± 0.89% and a limit of quantitation (LOQ) of 5.4% ± 3.1%24. The standard curve may saturate for Pra percentages >50%. Please click here to view a larger version of this figure.
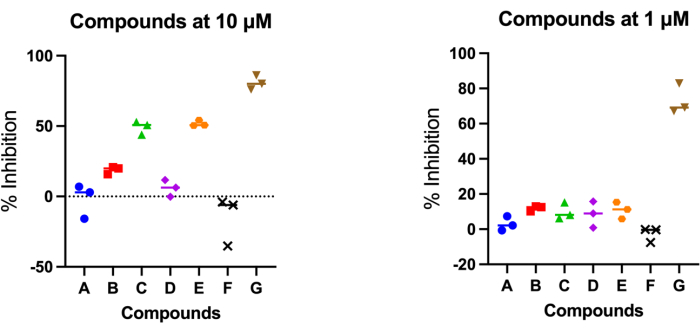
Figure 3: Testing new compounds in triplicate. Acetyl-click assay was used to assess the inhibitory activity of newly synthesized compounds. Compounds were diluted to 10 µM and 1 µM for testing in triplicate, with median indicated. Please click here to view a larger version of this figure.
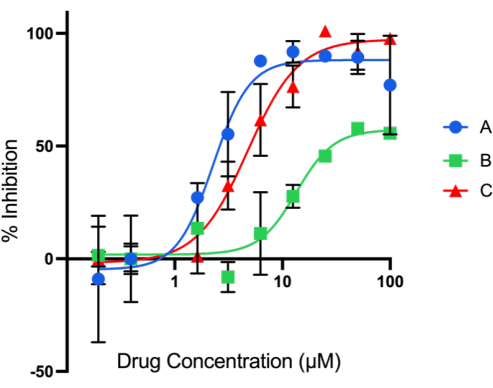
Figure 4: Serial dilution of test compounds in HAT1 acetyl click assay. Compounds were diluted from 100 µM to 0.01 µM and tested in duplicate. The resulting dose curves show that compounds A and C inhibit HAT1 activity by nearly 100% for several dilutions. Data were then used to calculate IC50 of compounds to be used in further experiments. Data are mean ± SEM of duplicate experiments. R2 values for nonlinear fits are 0.84, 0.78, and 0.94 for compounds A, B, and C, respectively. Please click here to view a larger version of this figure.
| % Positive Control | Positive Control Peptide [2.5 rxns] “pra” diluted to 0.1 mg/mL in DMSO (alkyne-containing) | H4 Peptide [2.5 rxns] diluted to 0.1 mg/mL in DMSO |
| 100 | 2.5 µL | - |
| 75 | 1.875 µL | 0.625 µL |
| 50 | 1.25 µL | 1.25 µL |
| 25 | 0.625 µL | 1.875 µL |
| 10 | 1 µL | 9 µL |
| 5 | 1.25 µL (of 1:10) | 1.25 µL |
| 1 | 1 µL (of 1:10) | 9 µL |
| 0 | - | 2.5 µL |
| Total | 8.5 µL | 25.5 µL |
Table 1: Dilution of Pra control peptide to H4 peptide. Ratios of positive control "Pra" peptide to H4 peptide that are used to create a standard curve. After Pra stock is initially resuspended at 4 mg/mL in DMSO, it must be diluted 1:40 in DMSO to a final concentration of 0.1 mg/mL. Biotinylated H4 peptide must also be diluted to 0.1 mg/mL (34.8 µM) in DMSO. Then, each peptide should be mixed together in the shown proportions. Each standard will be added in duplicate to the 96-well plate. Fluorescence output values should be averaged together to create the standard curve.
