The flow of the experiment is briefly described in Figure 1. If all steps are completed successfully, the images obtained by the microscope should be equivalent to Figure 2: live MM cells should be clearly seen as bright disks and BMSCs or HUVECs should be barely visible and well distributed in the background. As seen in Figure 2A, MM cells range in size from one patient to the other, but in general they are smaller than cell lines. Since they practically do not replicate ex vivo during the interval of the experiment (96 hr) they can be seeded at higher concentrations (1-3 million cells/ml). Stroma should be barely detectable, as BMSC adhere to the bottom of the well and stretch, presenting a low profile. The human myeloma cell line H929 is significantly larger than patient-derived MM cells (Figure 2B), and replicates within 24 hr. To prevent crowding of the image, MM cell lines are seeded a lower densities, 0.3 million/ml. Figures 2C and 2D represent patient-derived MM cells and human myeloma cell lines H929, respectively, co-cultured with HUVECS. HUVECS first adhere to the bottom of the well and then begin to contact each other and form networks, which later become thicker and with lumens.
Figure 4 exemplifies what is to be expected from the processed videos generated by the image analysis algorithm, showing detected live MM cells pseudo-colored in green (Figure 4A) with no or very little green signal in BMSCs and HUVECs at the end of the experiment in the highest drug concentration, when all MM cells are dead (Figure 4B). The software should aggregate the results from different wells and build a chart depicting gradual loss of viability with time of exposure at different drug concentrations (Figure 4C). The curve of dose response of the control cell line (e.g., H929 or MM1.S) should be the same as for previous experiments; significant deviations might indicate problems with the stock solution of drug used in the experiment.
It is important to carefully observe all the wells seeded before starting the imaging process, as artifacts may mislead the image analysis software and lead to false results. For instance, Figure 5A shows the result of an excessive density of BMSCs in the well or too much time elapsed between trypsinization and seeding, leading to these cells forming clumps. Figure 5B shows “colonies” of MM cells. If cell lines are seeded at high densities, they will form colonies as they replicate and this reduces the accuracy of the digital image analysis algorithm as it becomes increasingly difficult to discern one cell from the other. Figure 5C is an example of the opposite, where too few MM cells were seeded. This often occurs when the number of MM cells obtained from the bone marrow aspirate is below 500,000, and the “dead volume” left in the tube before re-suspension becomes comparable to the volume of media required to re-suspend MM cells in high densities. To solve this problem, determine the dead volume for the tube being used and include it in the dilution calculations. Figure 5D is an example of skewed focal plane. Note how MM cells on top left are bright and the ones in the bottom right are dark. This is caused by am unevenly placed plate or incorrectly placed objective. This will cause MM cells in different sections of the image to be counted differently. Scale bars (bottom right of each image) represent 500 µm.
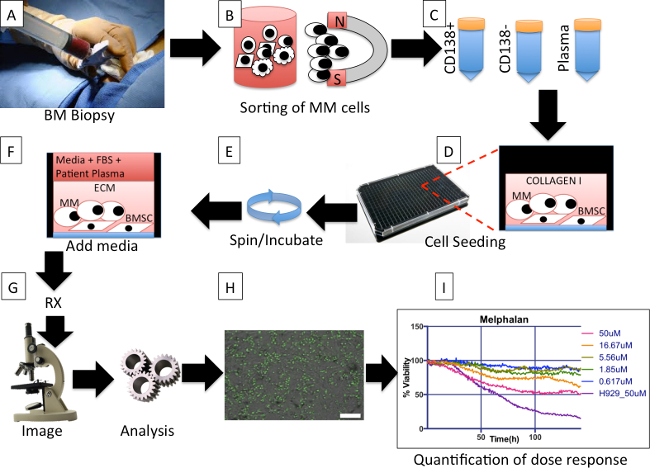
Figure 1. Protocol overall description. (A) During a bone marrow biopsy one extra tube of aspirate is obtained for the protocol. (B) Multiple myeloma (MM) cells are magnetically separated from other cells in the aspirate. (C) Three tubes, containing MM cells (MM), non-MM cells, and plasma, respectively, are obtained from the separation process. (D) Bone marrow derived mesenchymal stem cells (BMSCs) from previous aspirates are co-cultured with freshly obtained MM cells, and re-suspended in extracellular matrix (collagen or basement membrane matrix). (E) The plate is spun down to ensure that as many MM as possible are in the same focal plane and the BMSC are ready to adhere to the bottom of the well. The plate is placed in incubator until matrix polymerizes. (F) Each well is filled with a mix of culture media and patient-derived plasma. The plate is placed in an incubator O/N. (G) Drugs are added to each well and the plate is placed in a microscope equipped with a digital camera, motorized stage and bench top incubator, and imaged at regular intervals for a period of 96 hr. (H) The image files for each well are analyzed using an algorithm that segments live MM cells based on membrane motion (live cells are pseudo colored in green) and creates a spreadsheet with changes in cellularity for each well for each time point. Scale bar (bottom right) represents 500 µm. (I) A software program groups the wells by drugs and concentrations and creates a spreadsheet with curves of dose response normalized so that initial measures are 100%. Please click here to view a larger version of this figure.
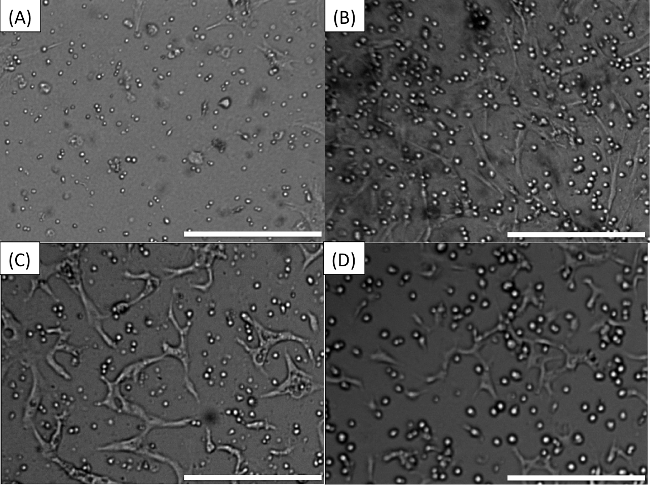
Figure 2. Pre-drug expected appearance of wells. (A) MM cells co-cultured with stroma. (B) The human myeloma cell line H929 co-cultured with stroma. (C) Patient-derived MM cells in co-culture with HUVECS. (D) Human myeloma cell line H929 in co-culture with HUVECS. Scale bars (bottom right of each Figure) represent 500 µm. Please click here to view a larger version of this figure.
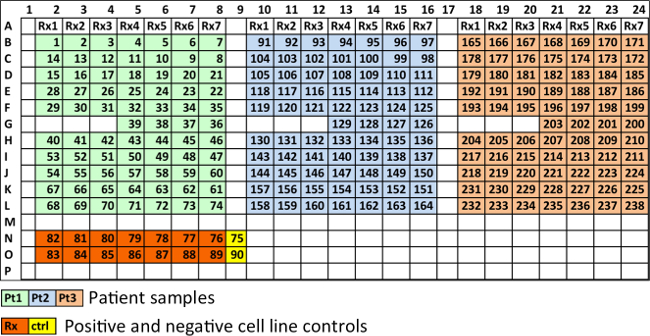
Figure 3. Recommended spatial distribution of wells in a 384-well plate. Each 384-well plate can carry 3 patient samples with 7 different drugs, or one patient sample tested with 21 drugs. Wells in light green, light blue and tan, contain MM cells from three different patients. Rx1, Rx2, etc., represent different drugs, each at five different concentrations and in duplicate. Four wells are used as controls for each patient (36-39 for patient 1, 126-129 for patient 2 and 200-203 for patient 3). Wells in orange contain cell lines at highest drug concentration of each drug, in duplicate. Wells in yellow are cell line controls (no drug). Please click here to view a larger version of this figure.
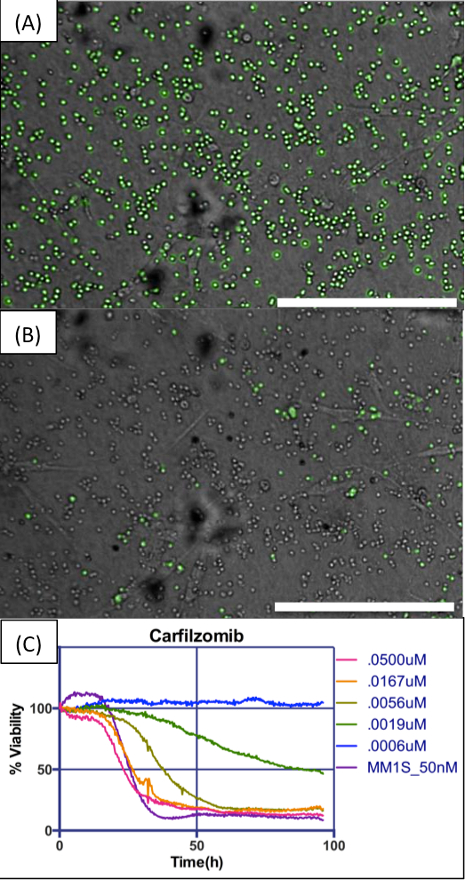
Figure 4. Example of digital image analysis and dose response charts. (A) MM cells in co-culture with BMSCs, embedded in collagen I, exposed to a concentration of 50 nM carfilzomib. Image analysis algorithm detects live cells and pseudo-colors them in green. (B) Images are acquired at regular intervals for the duration of the experiment. In this concentration of drug, almost all cells are dead at the end of 96 hr. (C) The chart shows changes in number of viable cells for five different concentrations of carfilzomib along an interval of 96 hr, normalized for time = 0 hr as 100%. The human myeloma cell line MM1.S was used as control for drug efficacy. Scale bars (bottom right of A and B) represent 500 µm. Please click here to view a larger version of this figure.
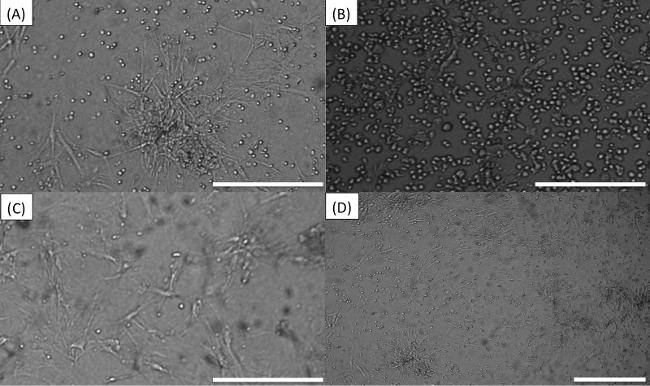
Figure 5. Common errors while seeding or imaging plate. (A) Stroma cells “tangled”. (B) “Colonies” of MM cells. (C) Too few MM cells. (D) Skewed focal plane. Scale bars (bottom right of each image) represent 500 µm. Please click here to view a larger version of this figure.
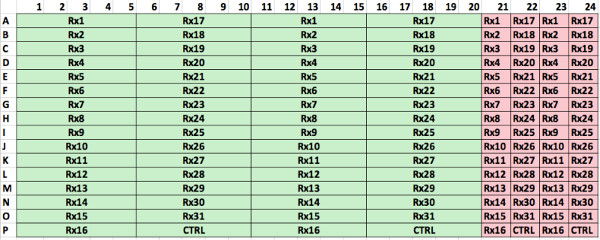
Supplemental Figure 1. Optimum spatial distribution of wells in 384-well plate using a robotic pipettor. In this configuration 31 different drugs can be tested at 5 different concentrations and 2 replicates (green). In addition, positive controls with a cell line can be used to ensure drug potency (pink). Please click here to view a larger version of this figure.
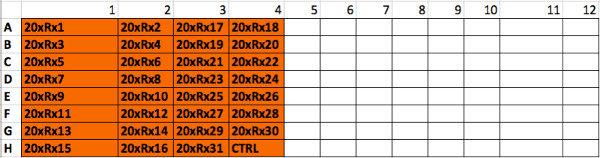
Supplemental Figure 2. Spatial distribution of drug “master plate”. The robotic pipettor will use this plate where each well contains one of the 31 drugs at 20x concentration, to create the drug dilution plate. Please click here to view a larger version of this figure.
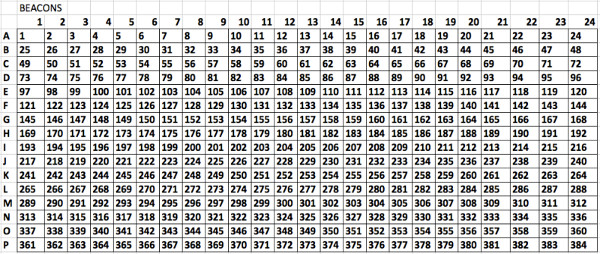
Supplemental Figure 3. Numbering of wells to be used when preparing a plate with a robotic pipettor. The software described in the protocol requires this numbering of the wells in order to adequately map the wells to the drugs and concentrations. Please click here to view a larger version of this figure.
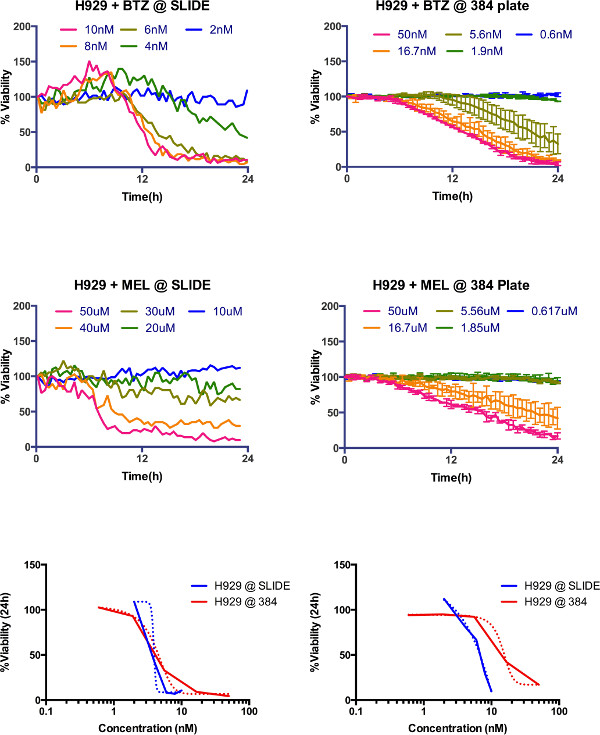
Supplemental Figure 4. Comparison between microfluidic slide (Khin et al., 2014) and current protocol. Dose response of H929 multiple myeloma cell line measured for 24 hr in the previous system (top left bortezomib, middle left melphalan) and in current system (top right bortezomib and middle right melphalan). For comparison purposes, LD50 at 24 hr for both platforms was 3.9 nM and 4.1 nM for bortezomib, and 6.4 µM and 14.65 µM for melphalan. Please click here to view a larger version of this figure.
