General Scheme of the Adult Zebrafish Neurosphere Culture
Here we describe all the steps of the protocol of a neurosphere-forming assay performed from the adult zebrafish brain. First, adult neural stem/progenitor cells have been collected either from zebrafish whole brain or from several dissected brain regions such as the telencephalon, the tectum and the cerebellum (Figures 1A-C). Single cell suspension of adult neural stem/progenitor cells have then been used to generate floating and self-renewing neurospheres (Figures 1D, E). Finally, neurospheres have been instrumental in studying the down- and up-regulation of the expression of coding genes or small non-coding RNAs11 in order to investigate their roles in the proliferation and differentiation of zebrafish neural stem/progenitor cells.
Neurosphere Passaging
Zebrafish primary neurospheres can self-renew following several passages. To generate neurospheres at Passage 1 and 2, steps 5.1-5.4 were repeated twice, in a total of 6-8 days. From DiV2-4, the size of the neurospheres increased up to around 50 μm in diameter. Secondary and tertiary neurospheres were also obtained after Passage 1 and 2, respectively (Figure 2D). At Passage 3, zebrafish neurospheres were however unable to grow up at the critical size of 50 μm diameter and failed to self-renew, suggesting that our culture condition rather selects a pool of stem/progenitor cells than a pure population of neural stem cells4.
Neurosphere Differentiation
Primary, secondary or tertiary neurospheres derived from either the whole brain or from the telencephalic, cerebellar and tectal area of adult zebrafish were tested for their differentiation potentialities. Between 1 and 3 days in vitro in differentiation conditions (DiVd1-DiVd3), a monolayer of adherent cells was observed (Figures 3A, B). As illustrated in Figure 3C, at DiVd4, axonal like projections as well as glia cells were visible and distinguishable by immunohistological or gene expression analyses11, assessing that neural stem/progenitor cells had differentiated. Similarly, neurons and glial cells differentiated from neural stem/progenitor cells isolated from different brain territories (Figures 3D, E).
Gene Expression Manipulation in Zebrafish Neurospheres
We have tested the role of miR-107 on the neuronal and glial differentiation of whole zebrafish brain-derived neural stem/progenitor cells (Figure 4). We showed that the downregulation of miR-107 by anti-miR-107 did not affect neurosphere formation but altered neuronal differentiation, as indicated by the abnormal growth of axonal processes (Figures 4A, B). RT-PCR gene expression analyses confirmed that inhibition of miR-107 leads to an increased expression of both the neuroblast marker Neurogenin-1 (Ngn-1) and axon-specific molecules, such as Map-2 and α-tubulin, without affecting glial cell marker expression (S-100, GFAP, Olig2) (Figure 4C). Accordingly, the gain of miR-107 expression by miR-107-mimic induced a decrease of neuroblast- and axon-specific marker expression (Figure 4D), assessing that, in vitro, miR-107 acts as a neuronal differentiation regulator during zebrafish neurogenesis11.
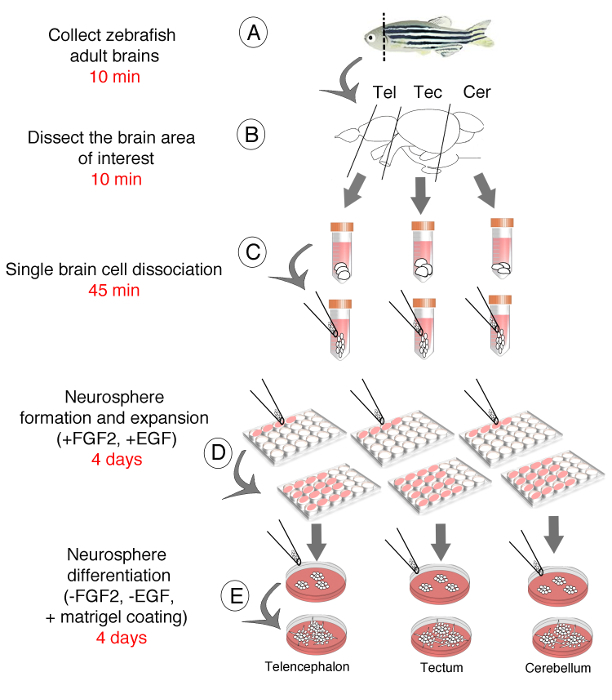
Figure 1: Schematic Representation of Protocol Steps. The procedures and associated timing include the dissection of either the whole brain or the telencephalic, tectal and cerebellar regions of the adult zebrafish brain (A, B), the obtention of a single cell suspension (C), the generation of floating neurospheres (D) and the differentiation of neurospheres (E).
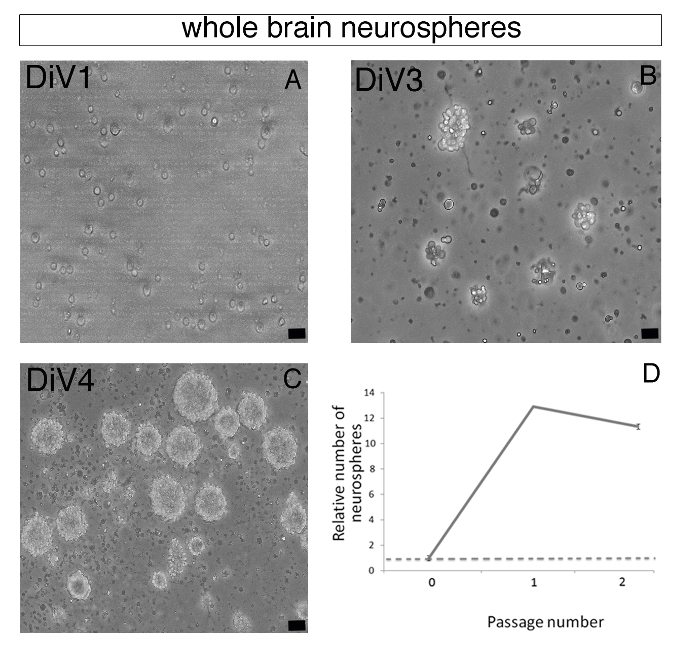
Figure 2: Forming Zebrafish Brain-derived Neurospheres. (A–C) Representative phase contrast images of whole brain-derived primary floating neurospheres observed at day 1 (DiV1, A), day 3 (DiV3, B) and day 4 (DiV4, C). (D) Chart representing the relative number of neurospheres at Passages 1 and 2 compared to the neurosphere number at Passage 0. After Passage 2, neurospheres formation was drastically decreased. Scale bar: 25 µm.
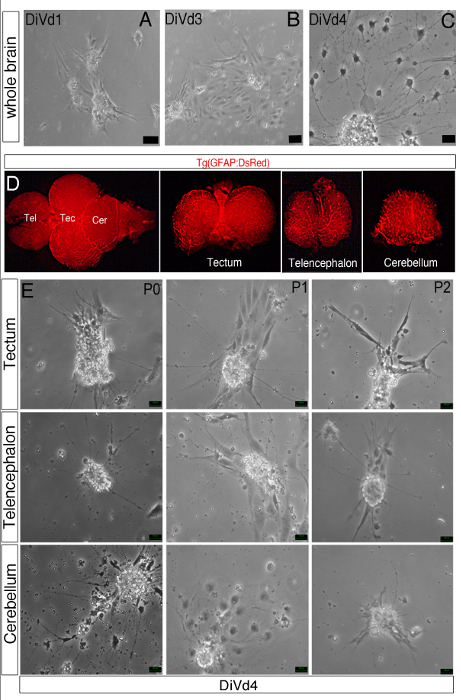
Figure 3: Differentiation of Zebrafish Brain-derived Neurospheres. (A–C, E) Phase contrast images of whole brain-derived neurospheres cultured in the Z-differentiation medium during 1 (A, DiVd1), 3 (B, DiVd3) and 4 (C and D, DiVd4) days. (E) Dorsal view of the whole brain of a 12 month old Tg(GFAP:DsRed) zebrafish. The telencephalon (Tel), tectum (Tec) and cerebellum (Cer) were dissected and collected as shown. (D) Images of DiVd4 neurospheres derived from the tectum, telencephalon and cerebellum at Passage 0 (P0), 1 (P1) and 2 (P2). Scale bar: 25 µm.
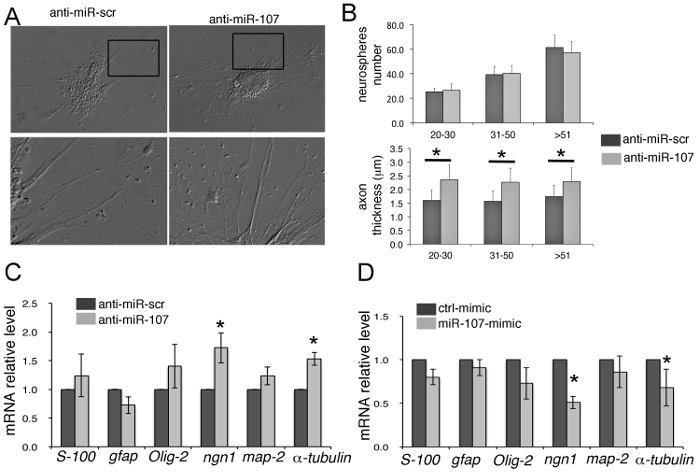
Figure 4: Analysis of Neurospheres Formed following MiR107 Manipulation. (A) Phase contrast images showing Div4d neurospheres treated at Div4 with the indicated oligonucleotides. Black boxes in the top panels indicate the area magnified in the bottom panels. (B) Top chart represents the quantification of the number of neurospheres formed with, or without, miR107. Neurospheres are subgrouped by their size (20-30 µm, 31-50 µm, >51 µm in diameter). Bottom chart indicates the quantification of the axonal projections from neurospheres treated and sub grouped as above. (C, D) qRT-PCR expression analysis of indicated genes by Div4d neurospheres previously treated with indicated oligonucleotides at Div4. Data represent the mean ± SEM, * p <0.05, n = 3. Scr: scramble. Scale bar: 25 µm.
| Problem | Possible reason | Solution |
| Recovery of too many dead cells | Time delay in preparing the brain before enzymatic treatment | Make sure the dissection set up and media are ready before sacrificing the fish (also check sterility, temperature) |
| Stringent papain treatment | Mechanical dissociation needs to be delicate enough to generate a live single cell suspension, but strong enough to avoid leaving behind too many clumps of tissue. Respect the recommended times and temperatures for incubation/centrifugation periods | |
| Neurospheres do not appear or grow poorly | Incorrect temperature | Check that incubator set at 30 °C is providing the correct temperature. |
| Growing spheres removed with debris | For each well, the aspiration of debris has to be performed well on top of the medium. Avoid entering into the medium with your pipet | |
| The integrity of some reagents (B27 supplement, EGF, FGF) is weakened | This integrity constitutes limiting factors in cell growth. Check batch number, and the way samples were preserved. Avoid thaw/refreeze cycles of any sampled reagent used in culture | |
| Presence of single cells | Mode of transfer/expansion of neurospheres is too harsh | This step is an expansion/dilution step because too many spheres are formed in the well. Avoid harsh sphere collection during transfer to prevent neurosphere dissociation into single cells |
| Absence of cells on matrigel | Matrigel was not properly thawed/diluted | Matrigel from -20 °C needs at least 30 min at 4 °C to thaw and remains viscous Pipet carefully |
| No adhesion on matrigel | The volume of cell suspension has to be small enough to allow cell deposition on matrigel coat | |
| Allow more time for cell deposition (up to 2 hr if larger volumes are used) | ||
| Fresh differentiation medium too cold | When replacing the cell-attachment medium, the differentiation medium needs to be warmed up at 30 °C to prevent thermal shock on the deposited cells | |
| Poor nucleofection | Dead cells | Make sure that you do not generate air bubbles while performing nucleofection. Use high quality DNA vectors by using Maxi-preparations. At least 1 – 2 hr after nucleofection, replace culture medium using either fresh Z-differentiation condition medium or Z-condition medium. |
| Few positive neurospheres | Neurospheres of varying sizes can lead to poor nucleofection. Using neurospheres at DiV2 and DiV3 are ideal for nucleofection. Density of cells should not exceed 4 x 103 neurospheres ml-1 in a 100 ml reaction. |
Table 1: Troubleshooting Advice Listing, for Each Step of the Protocol, the Possible Issues and the Solutions to Overcome Them.
