The Ex Vivo Culture and Pattern Recognition Receptor Stimulation of Mouse Intestinal Organoids
Summary
Here, a protocol to harvest, maintain, and treat mouse small intestinal organoids with pathogen associated molecular patterns (PAMPs) and Listeria monocytogenes is described, as well as emphasis on gene expression and proper normalization techniques for protein.
Abstract
Primary intestinal organoids are a valuable model system that has the potential to significantly impact the field of mucosal immunology. However, the complexities of the organoid growth characteristics carry significant caveats for the investigator. Specifically, the growth patterns of each individual organoid are highly variable and create a heterogeneous population of epithelial cells in culture. With such caveats, common tissue culture practices cannot be simply applied to the organoid system due to the complexity of the cellular structure. Counting and plating based solely on cell number, which is common for individually separated cells, such as cell lines, is not a reliable method for organoids unless some normalization technique is applied. Normalizing to total protein content is made complex due to the resident protein matrix. These characteristics in terms of cell number, shape and cell type should be taken into consideration when evaluating secreted contents from the organoid mass. This protocol has been generated to outline a simple procedure to culture and treat small intestinal organoids with microbial pathogens and pathogen associated molecular patterns (PAMPs). It also emphasizes the normalization techniques that should be applied when protein analysis are conducted after such a challenge.
Introduction
The ability to harvest and culture primary organoids have been described for small intestine, colon, pancreas, liver and brain and are exciting advances germane to understanding a more physiologically representative phenomena for tissue biology1-5. The first methods describing the culture and maintenance of small intestinal organoids was reported by Sato et al. out of the lab of Hans Clevers1. Prior to this method, harvesting and culture of primary intestinal epithelial cells proved to be limited and ineffective in sustaining epithelial cell growth. Methods included dissociation of tissue via incubation with enzymes, such as collagenase and dispase, which would ultimately lead to the outgrowth of intermixed primary fibroblast cells6. These conditions would also be time restricted in sustaining the epithelial cell culture. Minimal to no epithelial cell niche would form, as the epithelial cells would enter apoptosis due to the lack of appropriate growth factors or loss of contact integrity, termed anokis7. The advent of the 3D-organoid culture system has provided a method to culture primary intestinal cells containing a spectrum of intestinal cell types in sustained culture1. These epithelial organoids have advantages over cell lines being that they are composed of several differentiated cells, and better mimic the organ they are derived from in vivo8. The process to ultimately "grow a mini gut in a dish" has proven to be a valuable tool for assessing the response of intestinal epithelium under different stimuli. Investigating the interaction of primary intestinal cells with microbial pathogen associated molecular patterns (PAMPs) is relevant to the field of immunology as these molecular patterns can regulate diverse responses from both host and microbe9. Not only can investigators now explore these interactions with mouse organoids, but they can be cultured from humans as well2. This technology has the potential to dramatically alter personalized medicine and it is tempting to speculate about advances that this technique will make possible in the near future.
The overall goal of this method is to provide a protocol for the culture, expansion, and treatment of intestinal organoids with a variety of stimuli. Such stimuli can ultimately range from vaccines, bacterial PAMPs, live pathogens, gastrointestinal (GI) and cancer therapeutics. The isolation and culture of mouse intestinal organoids has been adapted from Sato et al. Though there are slight deviations from the original method, the end product being organoid culture is still achieved when following this protocol. This method is focused on describing an adequate technique for proper normalization when working with non-homogenous cell structures, which must be taken into consideration when conducting an assay based on cell number.
Protocol
All research was approved and conducted under Virginia Tech IACUC guidelines
1. Prepare R-Spondin1 Conditioned Media From HEK293T-Rspo1 Cell Line
- Generation of HEK293T-Rspondin1 cells has been previously described10. Seed HEK293T-Rspondin1 secreting cells at 5-10% confluency, approximately 8 x 105-1.7 x 106 cells, in a T-175 flask with 40 ml of 1x Dulbecco's Modified Eagle Medium (DMEM) + 10% Fetal Bovine Serum (FBS) as the growth media, and incubate at 37 °C + 5% CO2.
NOTE: The growth media for the HEK293T-Rspondin1 secreting cells will serve as R-spondin1 conditioned media. R-spondin1 can be alternatively purchased as a recombinant growth factor used at a final concentration of 500 ng/ml. - Grow the HEK293T-Rspondin1 cells for seven days or until reaching 95% confluency determined by bright-field microscopy. Harvest the 40 ml of conditioned media and transfer to a 50 ml conical tube. Discard the T-175 flask of HEK293T-Rspondin1 secreting cells.
- Centrifuge the tube containing conditioned media at 300 x g for 10 min at 4 °C to pellet cellular debris. Collect the supernatant, termed R-spondin1 conditioned media, and aliquot into 15 ml tubes. Store aliquots at -20 °C for short term or -80 °C for long term storage.
NOTE: Once thawed, the R-Spondin1 conditioned media can be stored up to 1 week a 4 °C.
2. Preparation of Organoid Growth Media and Reagents for Harvesting Small Intestinal Crypts
- One-day prior to harvesting, place the frozen protein matrix on ice and thaw overnight at 4 °C. Autoclave dissecting scissors, forceps and glass slides that will be used for crypt harvest.
- The following day, begin by preparing 40 ml of organoid growth media containing supplements and growth factors by adding stock concentrations to 40 ml of 1x DMEM/F-12. Media supplements contain: 10 mM2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) – 400 μl, 1x glutamine supplement – 400 μl, 1x B27 without vitamin A – 400 μl, 1x N2 – 200 μl, 1 mM N-acetyl-cysteine – 40 μl, 100 ng/ml m-Noggin – 40 μl, 50 ng/ml m-EGF – 4 μl and place in a 37 oC water bath until usage. Warm a 15 ml aliquot of R-spondin1 to 37 °C in a water bath.
- Warm three sterile 24 well plates to 37 °C for at least 30 min by placing in an incubator. Prepare 50 ml per mouse 1x Phosphate Buffered Saline (PBS) + 2 mM Ethylenediamine tetraacetic acid (EDTA) and cool to 4 °C. Prepare 100 ml per mouse 1x PBS containing 10% FBS and cool to 4 °C.
3. Harvesting Mus musculus Small Intestinal Crypts for Organoid Culture1
- Sacrifice a male C57B6/J mouse age 6-12 weeks of age raised on standard rodent chow and water available ad libitum according to institutional guidelines. Euthanize via CO2 asphyxiation followed by cervical dislocation.
NOTE: Keep carcass on ice until tissue harvest and perform the tissue harvest in a biological safety cabinet to minimize the risk of contamination.
Note: A male or female mouse may be used to generate organoids. - (Optional) Shave the mouse to remove the fur before submersing in 70% ethanol. Submerse the mouse in 70% ethanol for 2 min and pin limbs to pinning board with the dorsal side of the mouse touching the board.
- Use pre-sterilized dissecting scissors and forceps to make a mid-line incision on the ventral portion of the mouse. Make the incision at the genitalia proceeding cranial to the base of the neck. Open the skin incision laterally with tweezers and pin the skin to the pinning board to expose the peritoneum.
- Make a mid-line incision in the peritoneum of the abdomen with dissecting scissors and fold the peritoneum with forceps laterally and pin to the pinning board in order to expose the abdominal organs.
NOTE: Take care to avoid cutting abdominal organs. - Remove the small intestine from the abdominal cavity with dissecting forceps and scissors by cutting the proximal junction of the small intestine connected to the stomach and the distal junction connected to the caecum. Place the small intestine in a sterile petri dish containing 10 ml of ice cold 1x PBS.
- Flush the contents contained within the lumen of the small intestine with ice cold 1x PBS using a 1 ml pipette. Repeat this step until the debris within the lumen of the small intestine is removed.
- Cut the small intestine with dissecting scissors into 3-4 approximately equal length strips and lay the strips longitudinally on a new sterile Petri dish. For each strip, insert the cutting edge of the dissecting scissors inside the lumen of the small intestine to make an incision the entire length of the strip. Use forceps to laterally fold open the incision in order to expose the lumen of the small intestine.
- Use a sterile glass slide to gently scrape away the villi on the luminal surface of the small intestine. Cut the small intestine into 1-2 cm length strips with dissecting scissors and transfer these strips to a 50 ml conical tube containing 10 ml ice cold 1x PBS.
- Mix the contents gently by inverting the 50 ml conical tube and allow the tissue contents to settle to the bottom of the 50 ml conical tube. Aspirate the 1x PBS and repeat this three times by washing the tissue with 10 ml of ice cold 1x PBS. On the final wash leave the conical tube on ice for 10 min containing 10 ml 1x PBS and the small intestine tissue segments.
- Aspirate the 1x PBS and add 25 ml of 1x PBS + 2 mM EDTA. Place on a rocking platform at 4 °C or in an ice bucket for 45 min. While the tissue is incubating, label six 15 ml conical tubes "fraction #1-6."
- After incubation, allow the tissue contents to settle to the bottom of the tube and aspirate 1x PBS + 2 mM EDTA. Add 10 ml of 1x PBS +10% FBS and shake the tube vigorously by hand 10 times.
- Allow the tissue contents to settle to the bottom of the tube. Remove and transfer the supernatant to the tube 15 ml conical labeled "fraction 1". Repeat the 1x PBS + 10% FBS shaking step (3.11) five times and transfer the supernatant contents in order to the appropriately labeled-fraction tube.
- Centrifuge at 125 x g for 5 min. Aspirate the supernatant and resuspend pellets in 5 ml pre-warmed 1x DMEM/F-12, with no growth factors added.
- Centrifuge at 78 x g for 2 min. Aspirate 4 ml of the supernatant and resuspend each pellet in 1 ml of remaining media.
- Remove 20 ml from each fraction and add to a glass slide. Visualize crypts and debris under a light microscope and determine which fractions to combine and pool accordingly to achieve the greatest percentage of crypts to debris ratio.
NOTE: Fractions 2-6 yield the greatest percentage of crypts to be plated. The fractions to pool are most often fraction 3 with fraction 4, and fraction 5 with fraction 6. - Centrifuge fractions to be plated at 125 x g for 5 min. Aspirate supernatant leaving 50-100 μl remaining above the pellet.
- Keep tubes on ice and add 1 ml of protein matrix to the pooled fractions. Pipet up and down slowly to prevent addition of air bubbles.
- Remove pre-warmed 24 well plates from 37 °C incubator and add 50 μl of protein matrix/crypt suspension in the middle of each well. Transfer the seeded-plate to a 37 °C incubator for 10 min to allow the protein matrix drop to solidify.
- Add 450 μl of previously prepared organoid growth media + 50 μl of R-spondin1 conditioned media per well. Incubate plates at 37 °C + 5% CO2 overnight.
- Add 50-100 μl of R-spondin1 conditioned media per well daily. Every 3rd day replace entire organoid growth media with freshly prepared organoid growth media 450 μl/well described in step 2.2. Additionally add 50-100 μl of R-spondin1 conditioned media per well.
NOTE: The protein matrix drop maintains its integrity for one week, but after 7 days proceed to passaging organoids.
4. Passaging Organoids Every 7th Day
- Thaw the protein matrix on ice overnight at 4 °C the day before passaging. Warm one sterile 24 well plate to 37 °C for at least 30 min. Prepare organoid growth media described in step 2.2 and keep at 37 °C until usage.
- Remove organoid plate from incubator and commence passaging of plate on ice. Aspirate growth media with a 10 ml pipette from 3-4 wells to start.
NOTE: Keep the aspirated media in the 10 ml pipette, as this will be used to dislodge the protein matrix drop in the next step. - In the aspirated-wells, pipette up and down in order to dislodge the protein matrix drop from the plate. Gentle scraping with the tip of a 10 ml pipette is helpful in dislodging the protein matrix drop. Transfer the contents to a 15 ml conical tube.
- Incubate the 15 ml conical tubes on ice for 10 min and centrifuge at 125 x g for 5 min at 4 °C. Observe a white pellet at the bottom of the conical tube. Gently aspirate and discard the majority of the supernatant/ old organoid growth media above the organoid pellet leaving 100-200 μl above the pellet.
- Break up the organoid crypts by using a 23 gauge needle attached to a 1 ml syringe. Aspirate and eject 10-15 times in order to dissociate the crypts. Minimize air bubble formation due to excessive pipetting.
- Resuspend the organoids in 500 μl of protein matrix and add 50 μl per well in a new pre-warmed 24 well plate. Add 450 μl of previously prepared organoid growth media + 50 μl of R-spondin1 conditioned media per well. Incubate plates at 37 °C overnight.
5. Plating Organoids on Day 14 for Pattern Recognition Receptor Stimulation with PAMPs and Listeria monocytogenes for Gene Expression Analysis
- Two days prior to challenge, streak out L. monocytogenes on a BHI agar plate.
- The following day pick a L. monocytogenes colony and grow in 10 ml of BHI broth at 37 °C overnight shaking at 200 rpm.
- Thaw the protein matrix on ice overnight at 4 °C the day before organoid challenge. Warm one sterile 24 well plate to 37 °C for at least 30 min. Prepare organoid growth media described in step 2.2 and keep at 37 °C until usage.
- Remove organoid culture from incubator and commence passaging of organoids.
- Aspirate media from 3-4 wells and keep the aspirated media in the 10 ml pipete as this will be used to dislodge the protein matrix drop. Pipette up and down in order to dislodge the protein matrix drop from the plate. Gentle scraping with the tip of a 10 ml pipette is helpful in dislodging the protein matrix drop. Transfer the contents to a 15 ml conical tube.
- Incubate the 15 ml conical tubes on ice for 10 min. Centrifuge at 125 x g for 10 min at 4 °C. Observe a white pellet at the bottom of the conical tube. Gently aspirate the supernatant leaving approximately 100 μl of residual supernatant behind.
- Resuspend the organoid pellet in 5 ml of ice cold 1x PBS and remove a 10 μl aliquot for counting. Add the 10 μl aliquot to the middle of a hemocytometer without the glass cover slip. Gently overlay the glass coverslip on top of the drop.
NOTE: The hemocytometer will clog with organoids if the glass slide is not first removed. - Count the total number of organoids via a light microscope in every grid/ the entire chamber of the hemocytometer and determine the organoid concentration: number of total organoids counted per 10 μl.
- Remove a appropriate volume from the 15 ml conical tube that will allow adequate numbers of organoids to be seeded for the assay and centrifuge this suspension at 125 x g for 10 min.
NOTE: The range between 40-100 organoids per well of a 24 well plate is sufficient for RNA analysis. The typical organoid size can range from 300 μm to 1,000 μm in diameter. - Aspirate the supernatant and resuspend the pellet in 500 ml of protein matrix without generating air bubbles. Add 50 ml of organoid/protein matrix suspension per well in a 24 well plate.
NOTE: The amount of protein matrix used to resuspend organoids will vary for the number of treatment conditions and technical replicates. - Add 500 μl of organoid growth media (without N-acetyl-cysteine) to each well and incubate at 37 °C + 5% CO2 for 1 hr.
- Prepare 2x concentrations of PAMPs (i.e. flagellin, lipopolysaccharide) and live L. monocytogenes. Measure the OD of live L. monocytogenes and steak on a BHI plate for counting. Treat organoids at 1 x 106 Colony Forming Units (CFU)/ml.
NOTE: The OD to CFU determination will vary by bacteria type and should be assessed prior to organoid stimulation. For example, an OD of 0.6 ≈ 1 x 109 CFU/ml for L. monocytogenes, but should also be assessed independently prior to the experiment. - Streak out a serial dilution from the treatment stock of L. monocytogenes to accurately determine the treatment CFU/ml. The authors find that a 1 x 108 dilution of 100 μl on a BHI agar plate is sufficient to count colonies to determine an accurate CFU/ml.
- Prepare a heat killed solution of L. monocytogenes by taking a 1 ml aliquot of the live L. monocytogenes solution and centrifuge at 5,000 x g for 10 min. Aspirate the supernatant and wash the bacterial pellet with 1 ml of 1x PBS.
- Centrifuge the L. monocytogenes at 5,000 x g for 10 min and resuspend the pellet in 1 ml of 1x PBS. Heat kill the L. monocytogenes in a 1.7 ml polypropylene tube by heating at 80-90 °C for 1 hr. Culture a 100 μl aliquot of the heat killed L. monocytogenes on a BHI agar plate to confirm no live bacteria remain.
- Add 500 μl of the appropriate 2x PAMP and/or microbe treatment to 500 μl resident media in designated wells determined by the user. Incubate at 37 °C + 5% CO2 for 24 hr.
NOTE: Adding a 500 μl aliquot of a 2x PAMP/microbe treatment to 500 μl of resident media will bring the treatment concentration to 1x in a total volume of 1 ml.
- The following day, manually count L. monocytogenes colonies for an accurate determination of treatment CFU/ml. Aspirate media from the plate of treated organoids and wash wells three times with 1x PBS.
- Proceed to RNA extraction via phenol/chloroform11 or a RNA extraction kit according to manufacturers instructions. Evaluate gene expression using standard qRT-PCR12.
6. Plating Organoids on Day 14 for PAMP and L. monocytogenes Challenge for Protein Analysis in Supernatant
- Two days prior to challenge, start a cell culture of Caco-2 cells, seeding density ≈1 x 106 cells in a T-75 flask with 1x DMEM + 20% FBS and incubate the Caco-2 cells at 37 °C + 5% CO2. Begin a bacterial culture of L. monocytogenes on BHI plate and incubate plate in a bacterial incubator at 37 °C.
- The following day pick a L. monocytogenes colony and grow in 10 ml of BHI broth at 37 °C overnight. Thaw the protein matrix on ice overnight at 4 °C the day before organoid challenge.
- The following day warm one sterile 96 well black-walled plate to 37 °C for at least 30 min. Prepare organoid growth media without N-acetyl-cysteine described in step 2.2 and keep at 37°C until usage. Remove organoid culture from incubator and commence passaging of organoids.
NOTE: The typical organoid size can range from 300 μm to 1,000 μm in diameter. - Aspirate media from 3-4 wells and keep the aspirated media in the 10 ml pipete as this will be used to dislodge the protein matrix drop. Resuspend with a pipette in order to dislodge the protein matrix drop from plate. Gentle scraping with a 10 ml pipette is helpful in dislodging the protein matrix drop. Transfer the contents to a 15 ml conical tube.
- Incubate the 15 ml conical tubes on ice for 10 min. Centrifuge at 125 x g for 10 min at 4 °C. Observe a white pellet at the bottom of the conical tube. Gently aspirate the supernatant leaving 100 μl of residual supernatant behind.
- Resuspend the organoid pellet in 5ml of ice cold 1x PBS and remove a 10 μl aliquot for counting. Add the 10 μl aliquot to the middle of a hemocytometer and gently overlay the glass coverslip.
NOTE: The hemocytometer will clog with organoids if the glass slide is not first removed. - Count the total number of organoids via a light microscope in every grid of the hemocytometer/ the entire chamber and determine the organoid concentration: number of total organoids counted per 10 ml.
- Remove a appropriate volume from the 15 ml conical tube that will allow adequate numbers of organoids to be seeded for the assay and centrifuge this suspension at 125 x g for 10 min.
NOTE: The amount of protein matrix used to resuspend organoids will vary for the amount of treatment conditions and technical replicates. The authors find that 40-100 organoids is sufficient for secreted protein analysis. - Aspirate the supernatant and resuspend the pellet in 500 μl of protein matrix /well without generating air bubbles.
NOTE: The amount of protein matrix used to resuspend organoids will vary for the amount of treatment conditions and technical replicates. - Leave at least two columns empty on the 96 well black-walled tissue culture plate as these will serve as wells seeded with Caco-2 cells for the generation of a standard curve. Add 50 μl of protein matrix + organoid suspension per well to a pre-warmed 96 well black-walled tissue culture plate. Store the plate at 37 °C for 10 min to allow protein matrix to solidify.
- Add 100 μl of organoid growth media (without N-acetyl-cysteine) to each well and incubate at 37 °C + 5% CO2 for 1 hr.
- Prepare 2x concentrations of PAMPs and live L. monocytogenes. Add 100 ml of each given treatment condition per well and incubate for 24 hr.
- The following day plate Caco-2 cells in the standard curve wells. Begin by warming sterile 1x PBS, 0.25% trypsin and 1x DMEM + 20% FBS to 37 °C.
- Remove the T-75 flask containing Caco-2 cells and aspirate the cell culture media. Wash the cells with 10 ml 1x PBS. Aspirate the 1x PBS, add 2 ml 0.25% Trypsin and place T-75 flask in 37 °C incubator for 15 min.
- Visualize the flask under a light microscope to ensure the cells have detached then add 8 ml of 1x DMEM + 20% FBS to the detached Caco-2 cells and transfer into a 15 ml conical tube. Remove a 10 μl aliquot and count the cells by light microscopy using a hemocytometer.
NOTE: An upper cell number of 60,000 cells/well works well and dilutions can be conducted for generation of the standard curve down to 2,500 cells/well. - Resuspend the Caco-2 cells in protein matrix and add 50 μl per well to appropriate wells. Allow the protein matrix to solidify at 37 °C for Caco-2 cells.
NOTE: Growth media for the Caco-2 cells does not need to be added following solidification of the protein matrix. - Following the 24 hr treatment time for the organoid assay, aspirate and save the organoid cellular supernatant media and keep on ice. Wash the wells three times with 1x PBS. Add 100 µl of 100% methanol to each well including the Caco-2 standard curve wells and incubate at 4 °C or on ice for 20-30 min to fix the organoids.
NOTE: It is not required that the Caco-2 standard curve wells be washed with 1x PBS, as they can have methanol added directly. - Following the methanol incubation, wash the wells three times with ice cold 1x PBS. Store the plate at 4 °C for up to 1-2 weeks.
- Add nuclear staining dye at a concentration of 1 µg/ml per well, cover the plate with aluminum foil and incubate at 4 °C overnight.
- Use a 96 well plate reader to measure nuclear staining dye excitation/emission (350nm/461nm respectively) and normalize each organoid well to the standard curve of Caco-2 cells.
- Proceed to downstream assays, such as ELISA12, of cell culture supernatant normalizing to cell number.
Representative Results
When following this protocol to cultivate intestinal organoids, characteristic sphere shaped organoids will be present after harvesting. The addition of R-spondin1 conditioned media daily will initiate the growth and budding of the organoids. The growth of organoids is shown in Figure 1A–F, and is representative of intestinal organoids on days 1, 2, 4, 5, 6 and day 14. Figure 1F represents the non-homogeneous growth characteristics of organoids on day 14.
Once the organoids are grown to an adequate number, they can be replated and challenged with various PAMPs and/or microbes. Expression analysis can be performed via standard techniques. This is represented in Figure 2A-C which shows the mRNA expression of inflammatory cytokines IL-18, IL-6, and TNFα which were analyzed following a 24 hr challenge of organoids with heat killed L. monocytogenes and the PAMP flagellin. The rationale for evaluating the mRNA expression cytokines IL-18, IL-6, and TNFα was that these were good candidates for being modulated by epithelial cells in response to pathogenic challenge, and modulation of these inflammatory cytokines would demonstrate the effectiveness of the technique.
Figure 3A–C demonstrates the relative nuclear staining of intestinal organoids with nuclear staining dye following fixation with minimal background staining of debris in the resident protein matrix. This method of fixation and staining can be applied to normalize assays, which will account for differing cell numbers in each well. This is apparent in Figure 4 when generating a standard curve of serial dilutions of Caco-2 cells plated in protein matrix, then fixed and stained with nuclear staining dye. The R2 value of 0.89 indicates a linear relationship between cell number and mean fluorescent intensity, and the linear equation can be used to normalize organoids to cell number. The standard curve depicted in Figure 4 begins to reach the saturation limit beyond 30,000 cells per well. A titration of cells above 30,000 cells per well has been shown in Figure 4 to include the saturation range of nuclear staining dye. Increased accuracy of normalization will be obtained with a cell number that reflects the linear range of the nuclear staining dye before the saturation limit is reached.
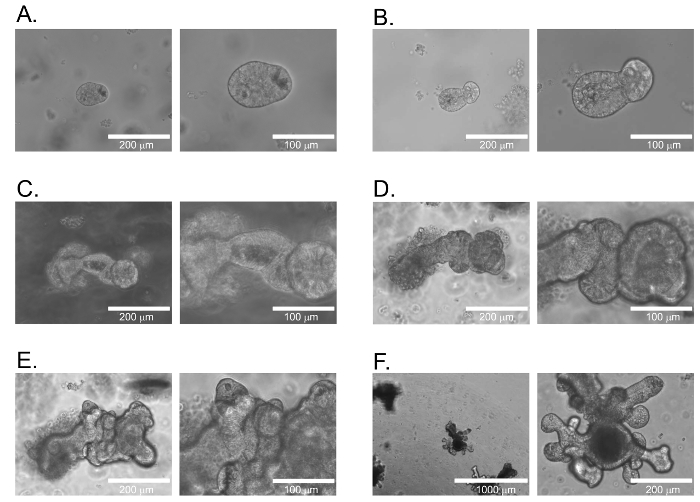
Figure 1: Small Intestinal Organoid Growth. Time course of growth for murine small intestine derived organoids following isolation. (A) Day 1. (B) Day 2. (C) Day 4. (D) Day 5. (E) Day 6. (F) Day 14. Images are representative of organoid growth for each given day. Scale bars equal 200 μm in the left image for A, B, C, D, and E. Scale bars equal 100 μm in the right image for A, B, C, D, and E. Scale bars equal 1,000 μm and 200 μm for left and right images for F, respectively. Please click here to view a larger version of this figure.
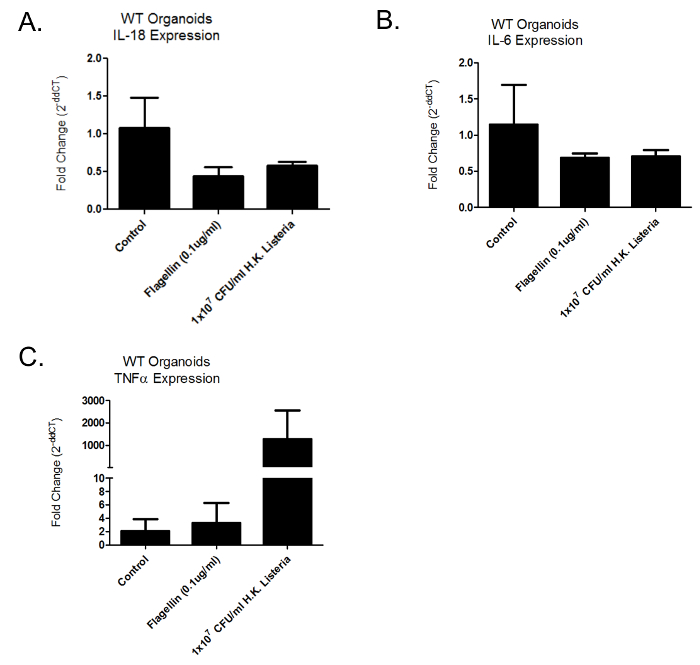
Figure 2: mRNA Expression of Inflammatory Cytokines Following 24 hr PAMP Challenge. Relative mRNA expression of wild-type intestinal organoids challenged for 24 hr with PAMPs and heat killed Listeria monocytogenes. (A–C) represent fold change of the inflammatory cytokines IL-18, IL-6 and TNFα respectively. Error bars represent Standard Deviation (S.D.) Please click here to view a larger version of this figure.
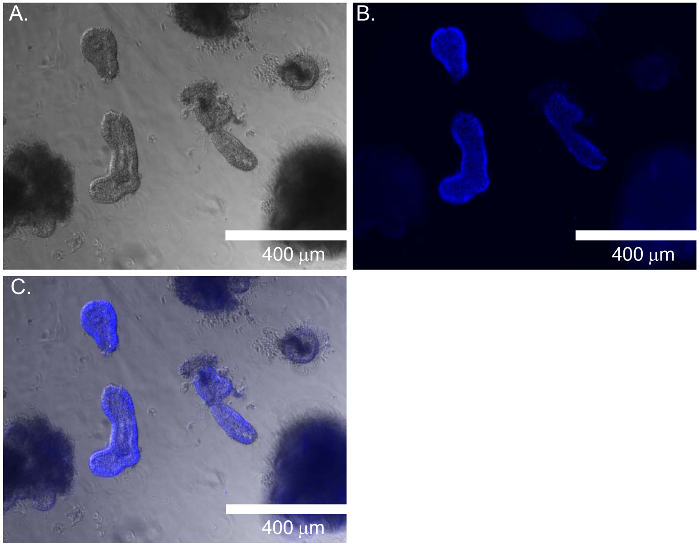
Figure 3: Relative Fluorescent Staining of Organoids for Normalization. Nuclear staining of organoids with following fixation. (A) Bright field of organoids. (B) Fluorescent staining of organoids with nuclear staining dye. (C) Merged image of bright field and fluorescent staining. Scale bars equal 400 μm in all images. Please click here to view a larger version of this figure.
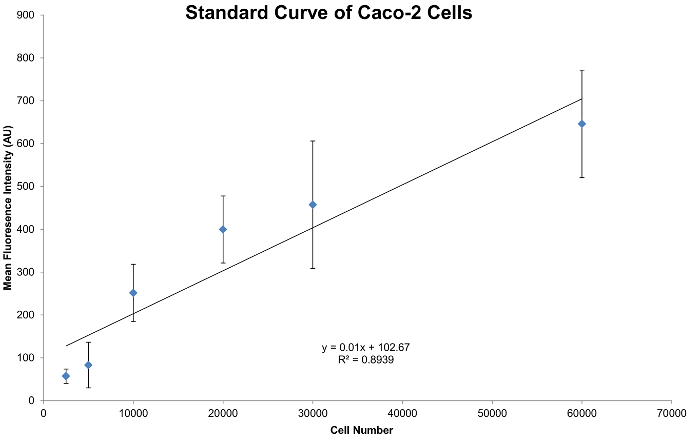
Figure 4: Standard Curve generated from Caco-2 Cells. Standard curve generated from triplicate wells. Cell seeding had a range starting at 60,000 cells per well with dilutions down to 2,500 cells per well. Please click here to view a larger version of this figure.
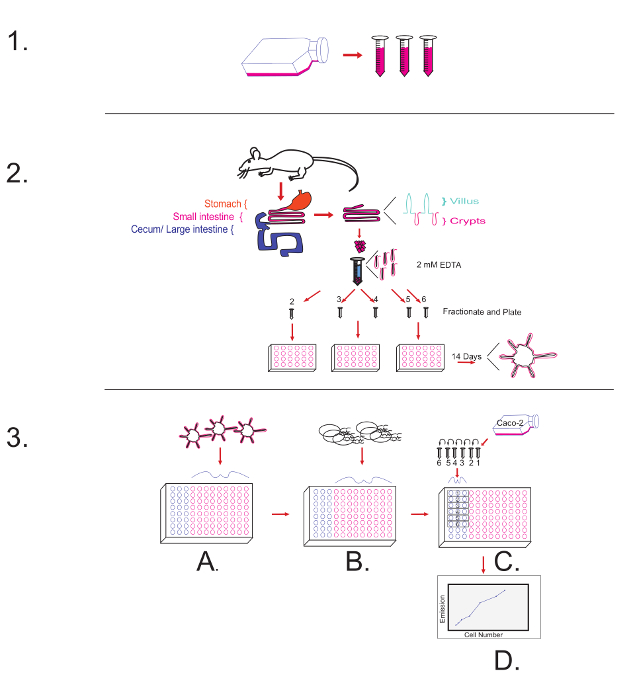
Figure 5: Overview of the Protocol Diagram. 1-Top Panel of the Figure illustrates harvesting the R-spondin1 conditioned media. 2-Middle Panel of the Figure illustrates aspects of protocol to harvest and culture mouse small intestinal organoids. 3-Bottom Panel of the Figure illustrates: (A) The plating of 14 day cultured organoids. (B) Challenge with PAMPs/ L. monocytogenes, and leaving unoccupied wells to generate a standard curve. (C) Following the PAMP challenge, the plating of Caco-2 cells in the previously unoccupied wells to generate a standard curve. (D) Following the fixation and addition of a nuclear staining dye, measuring the excitation/ emission of the wells and normalizing against a standard curve. Please click here to view a larger version of this figure.
Discussion
The culture and maintenance of intestinal organoids is a procedure that can be mastered by any individual with adequate tissue culture technique. There are subtleties in passaging when compared to growing cells in a more conventional monolayer, but these subtleties are not difficult to overcome. The critical steps of this method involve being able to grow the organoids to a high enough density for optimal seeding. Experiments must be scaled down with organoids as large seeding densities that can commonly be achieved with cell lines are not practical. This becomes especially apparent when there are multiple treatment groups.
This protocol is intended to provide a step-by-step method to study host-pathogen interactions of the intestinal epithelium with a variety of different bacterial, viral, and fungal pathogens, as well as address difficulties using this system with respect to normalization. Reports are available that describe the interactions of intestinal organoids with the bacterial pathogen Salmonella, yet do not address normalization methods when measuring secreted cytokines13.
There are several difficulties that are encountered when normalizing organoid cultures by different methods. Normalizing secreted protein via bicinchoninic acid assay (BCA) is an option; however, the growth components required for organoid culture (N2 and/or Vitamin B27) interfere with the BCA assay (data not shown). Normalizing via cellular viability, such as a modified MTT assay has been described14; however, a treatment that will alter the mitochondrial metabolic activity of the organoids will introduce an inaccurate method for normalization via this technique as MTT is based on reduction by the action of mitochondrial dehydrogenases15. It is also necessary to remove N-acetyl-cysteine (NAC) from the media, not only if normalization via the MTT technique is desired, but if treating with a bacterial pathogen as NAC can inhibit bacterial growth16.
Advantages of this technique are that secreted cytokines and protein products from organoids can now be normalized against a standard curve of Caco-2 cells. Limitations of this technique are that the normalization is correlative because nuclear staining of non-transformed primary epithelial organoids are normalized against the nuclear staining of a colon cancer cell line. The authors find that fixing with methanol and staining nuclei with nuclear staining dye is an effective normalization technique against a standard curve of Caco-2 cells. Although the growth characteristics of this colon cancer cell line do not exactly mimic the growth characteristics of intestinal organoids, using a nuclear stain and measuring mean fluorescence intensity is a good strategy to normalize against total cell number.
Taken together, the technique described here provides a good starting point to mimic host-pathogen interactions with adequate normalization that is essential for making accurate interpretations using this model system. The significance of this technique with respect to alternative methods is that proper normalization must be performed when conducting any evaluation of secreted protein, such as ELISA based assays.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors would like to thank Dr. Sheryl Coutermarsh-Ott, Dylan McDaniel and Bettina Heid for technical discussions. The authors thank Dr. Nanda Nanthakumar for providing the Caco-2 cells. The authors also thank The Multicultural Academic Opportunities Program (MAOP) at Virginia Tech. This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases Award K01DK092355 (to I.C.A.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Materials
| Fetal Bovine Serum (FBS) | Atlanta Biologicals | S11050 | (Section 1,3,6) Or equivalent brand |
| Sorvall Legend XTR Centrifuge | Thermo | (Section 1,3) | |
| DMEM | GE Healthcare | Sh30243.01 | (Section 1,6) For Caco-2 and HEK293 Rspondin1 cells |
| HEK293T-Rspondin1 secreting cell line | (Section 1) Described and modified from Kim, K.A. et al. Lentiviral particles contained RSPO1(NM_138683) ORF cDNA cloned into a pReceiver-Lv105 backbone custom ordered and purchased from GeneCopoeia. | ||
| 50 ml conical tube | Falcon | 352070 | (Section 1) Or equivalent brand |
| T-175 Flask | Corning | 431079 | (Section 1) Or equivalent brand |
| Protein Matrix | Corning | 356231 | (Section 2,3,4,5,6) Matrigel Growth Factor Reduced |
| HyClone Dulbecco's (DPBS) | GE Healthcare | SH30264.01 | (Section 2,3) |
| DMEM/F12 | Life Technologies | 12634-010 | (Section 2,3) Advanced DMEM/F12 |
| Corning 24 Well TC Plates | Corning | 3524 | (Section 2) |
| N2 Supplement 100x | Life Technologies | 17502-048 | (Section 2) |
| B27 without vitamin A 50x | Life Technologies | 12587-010 | (Section 2) |
| Trizol | Life Technologies | 15596-026 | (Section 2) |
| Glutamine Supplement (Glutamax) | Life Technologies | 35050-061 | (Section 2) Can Combine with Advanced DMEM/ F12 |
| HEPES (1 M) | Life Technologies | 15630-080 | (Section 2) Can Combine with Advanced DMEM/ F12 |
| 10ml Serological Pipet | Falcon | 357551 | (Section 2) Or equivalent brand |
| Murine Noggin | Peprotech | 250-38 | (Section 2) Stock = 100 mg/ml |
| N-Acetyl-L-cysteine | Sigma-Aldrich | A9165 | (Section 2) Stock = 1M |
| Recombinant Mouse EGF | Biolegend | 585608 | (Section 2) Stock = 500 mg/ml |
| Rocker Variable | Bioexpres | (Section 3) | |
| dissecting scissors | (Section 3) | ||
| forceps | (Section 3) | ||
| glass slides | (Section 3) | ||
| dissecting tweezers | (Section 3) | ||
| 25 ml Serological Pipet | Falcon | (Section 3) | |
| EDTA | Sigma-Aldrich | SLBB9821 | (Section 3) 0.5M or alternative TC grade EDTA |
| Sterile Petri Dish 100mm x 15mm | Fisher | FB0875712 | (Section 3) Or equal sized TC dish |
| 1ml Syringe | Becton Dickinson | 309659 | (Section 4) |
| Precision Glide Needle | Becton Dickinson | 305120 | (Section 4) 23G x 1 1/4 (0.6mm x 30mm) |
| Flagellin from Bacillus subtilis | Invivogen | tlrl-bsfla | (Section 5,6) |
| Listeria monocytogenes | ATCC | 19115 | (Section 5,6) (Murray et al.) |
| Hemocytometer | Sigma-Aldrich | Z359629-1EA | (Section 5,6)Or equivalent brand |
| BBL Brain Heart Infusion Agar | Becton Dickinson | 211065 | (Section 5) |
| Bacto Brain Heart Infusion | Becton Dickinson | 237500 | (Section 5) |
| Caco-2 | ATCC | HTB-37 | (Section 6) |
| Trypsin | gibco | 25200056 | (section 6) |
| Methanol | Fisher | A412-4 | (Section 6) |
| SpectraMax M5 | Molecuar Devices | (Section 6) | |
| 96 Well Assay Plate | Corning | 3603 | (Section 6) Black Plate, Clear Bottom TC treated |
| Nuclear Staining Dye | Life Technologies | H1399 | (section 6) Hoechst 33342 |
| T-75 Flask | Corning | 430641 | (Section 6) Or equivalent brand |
| 15 ml conical tube | Falcon | 352096 | (Section1,3) Or equivalent brand |
| 1.7 ml polypropylene tube | Bioexpress | C-3262-1 | Or equivalent brand |
| Quick-RNA MiniPrep | Zymo Research | R1054 | Or equivalent brand |
| TNF-alpha | Applied Biosystems | Mm 00443260_g1 | Taqman gene expression assay kit |
| IL-6 | Applied Biosystems | (Mm 00446190_m1 | Taqman gene expression assay kit |
| IL-1beta | Applied Biosystems | Mm 00434228_m1 | Taqman gene expression assay kit |
| IL-18 | Applied Biosystems | Mm 00434225_m1 | Taqman gene expression assay kit |
| 18s | Applied Biosystems | Hs 99999901_s1 | Taqman gene expression assay kit |
| 7500 Fast Real Time PCR System | Applied Biosystems | ||
| Nexus gradient Mastercycler | Eppendorf | ||
| TaqMan Fast Universal PCR Master Mix | Life Technologies | 4352042 | |
| High Capacity cDNA Reverse Transcription Kit | Life Technologies/Applied Biosystems | 4368814 | |
| Fast Optical 96-Well Reaction Plate, 0.1 mL | Life Technologies/Applied Biosystems | 4346907 | |
| Recombinant Mouse R-Spondin 1 Protein | R&D Systems | 3474-RS-050 | 500 ng/ml |
| chloroform | Sigma-Aldrich | C7559 |
References
- Sato, T., et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 459 (7244), 262-265 (2009).
- Sato, T., et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 141 (5), 1762-1772 (2011).
- Boj, S. F., et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 160 (1-2), 324-338 (2015).
- Huch, M., et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell. 160 (1-2), 299-312 (2015).
- Lancaster, M. A., et al. Cerebral organoids model human brain development and microcephaly. Nature. 501 (7467), 373-379 (2013).
- Freshney, R. I., Freshney, M. G. . Culture of epithelial cells. 2nd edn. , (2002).
- Vachon, P. H., et al. Differentiation state-selective roles of p38 isoforms in human intestinal epithelial cell anoikis. Gastroenterology. 123 (6), 1980-1991 (2002).
- Hynds, R. E., Giangreco, A. Concise review: the relevance of human stem cell-derived organoid models for epithelial translational medicine. Stem Cells. 31 (3), 417-422 (2013).
- Kaiko, G. E., Stappenbeck, T. S. Host-microbe interactions shaping the gastrointestinal environment. Trends Immunol. 35 (11), 538-548 (2014).
- Kim, K. A., et al. Mitogenic influence of human R-spondin1 on the intestinal epithelium. Science. 309 (5738), 1256-1259 (2005).
- Chomczynski, P., Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat Protoc. 1 (2), 581-585 (2006).
- Allen, I. C., et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity. 34 (6), 854-865 (2011).
- Zhang, Y. G., Wu, S., Xia, Y., Sun, J. Salmonella-infected crypt-derived intestinal organoid culture system for host-bacterial interactions. Physiol Rep. 2 (9), (2014).
- Grabinger, T., et al. Ex vivo culture of intestinal crypt organoids as a model system for assessing cell death induction in intestinal epithelial cells and enteropathy. Cell Death Dis. 5, 1228 (2014).
- Slater, T. F., Sawyer, B., Straeuli, U. Studies on Succinate-Tetrazolium Reductase Systems. Iii. Points of Coupling of Four Different Tetrazolium Salts. Biochim Biophys Acta. 77, 383-393 (1963).
- Parry, M. F., Neu, H. C. Effect of N-acetylcysteine on antibiotic activity and bacterial growth in vitro. J Clin Microbiol. 5 (1), 58-61 (1977).
