Quelle: Michael S. Lee1 und Tonya J. Webb1
1 Department of Microbiology and Immunology, University of Maryland School of Medicine and the Marlene and Stewart Greenebaum Comprehensive Cancer Center, Baltimore, Maryland 21201
Immunhistochemie (IHC) und Immunzytochemie (ICC) sind Techniken, die verwendet werden, um die Expression und Lokalisierung spezifischer Antigene mit Antikörpern zu visualisieren. Die erste veröffentlichte Anwendung von IHC war 1941, als Albert Coons die Technik benutzte, um das Vorhandensein von Pneumokokken-Antigen in Gewebeabschnitten von Mäusen zu visualisieren, die mit Pneumokokken infiziert waren (1). Der Name, Immunhistochemie, leitet sich von den Wurzeln “immuno-” in Bezug auf Antikörper und “Histo-” in Bezug auf die gewebeabschnitte ab, die in IHC verwendet werden. Die Wurzel “Cyto-” in der Immunzytochemie unterstreicht den entscheidenden Unterschied zwischen ICC und IHC. Während IHC Teile des gesamten Gewebes verwendet, verwendet ICC Zellen, die aus Gewebe isoliert oder in Kultur gewachsen sind. Der Unterschied in den verwendeten Proben bedeutet, dass die Probenvorbereitung technisch zwischen IHC und ICC unterschiedlich ist, aber ansonsten sind die Protokolle für ICC und IHC identisch und man wird feststellen, dass die Begriffe häufig austauschbar verwendet werden.
Sowohl bei IHC als auch bei ICC werden Antikörper mit chemischen oder fluoreszierenden Tags, wie Peroxidase bzw. Rhodalin, verwendet, um die Verteilung jedes Antigens von Interesse durch spezifische Bindung des markierten Antikörpers an das Antigen zu visualisieren. Im Falle von IHC werden dünne Gewebescheiben auf einem Dia immobilisiert, um die Struktur des Gewebes zu erhalten, bevor es gebeizt wird, was die Visualisierung von Antigenen im Kontext ganzer Gewebe ermöglicht (Abbildung 1). Im Falle von ICC werden Zellen gleichmäßig auf einem Dia verteilt, bevor sie gefärbt werden, was die Visualisierung der Antigenverteilung innerhalb einzelner Zellen ermöglicht, aber nicht innerhalb der Struktur eines bestimmten Gewebes. Aufgrund der Ähnlichkeiten zwischen den beiden Protokollen wird sich dieses Protokoll auf IHC konzentrieren, um die zusätzlichen Komplexitäten der Probenvorbereitung im Zusammenhang mit IHC zu beheben.
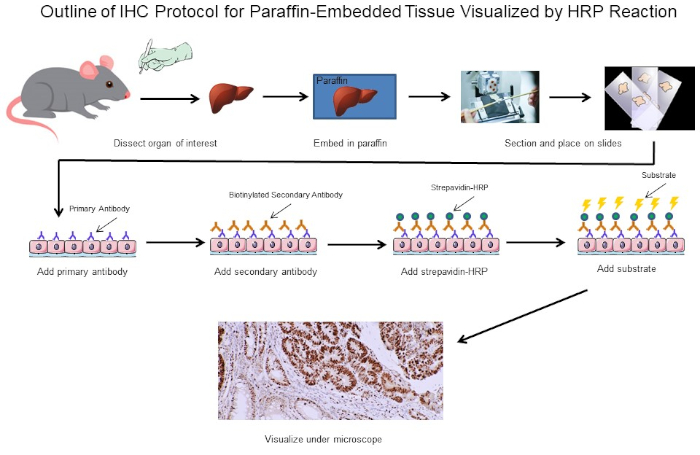
Abbildung 1: Gliederung des IHC-Protokolls. Visuelle Umriss eines IHC-Protokolls für paraffinintegriertes Gewebe, das von einer Maus seziert wurde. Dieses Protokoll verwendet einen biotinylierten Sekundärantikörper und Strepavidin-HRP, um die Position der Antikörperbindung zu visualisieren. Andere Optionen, wie fluoreszierend markierte Antikörper, sind ebenfalls möglich. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Die erste wichtige Entscheidung bei der Durchführung von IHC ist, wie die Gewebeabschnitte vorbereitet werden, um die Struktur des Gewebes während des gesamten Färbeprozesses zu erhalten. Die beiden wichtigsten Optionen sind formalin-feste Abschnitte von Paraffin-eingebettetem Gewebe oder frische Abschnitte von gefrorenem Gewebe. Es gibt keine einfache Antwort darauf, welche Methode verwendet werden soll, da es davon abhängt, welche nachgelagerte Analyse durchgeführt wird. Formalin-Fixierung von Paraffin eingebetteten Geweben wird im Allgemeinen gedacht, um Gewebemorphologie für eine optimale Bildgebung besser zu erhalten, während das Einfrieren von frischem Gewebe die Proteinfunktion für nachfolgende Assays außerhalb von IHC erhalten kann. Darüber hinaus haben sich frisch gefrorene Gewebeabschnitte als besser für die Genexpressionsanalyse geeignet erwiesen (2). Eine dritte Überlegung ist, ob die Antikörper für Ihr Antigen von Interesse für feste oder gefrorene Gewebeabschnitte geeignet sind, da einige Antikörper nur für eine bestimmte Art von Abschnitt optimiert wurden und möglicherweise nicht für andere arbeiten. Schließlich muss auch festgestellt werden, wie lange sie die Gewebeabschnitte lagern müssen, da frische gefrorene Proben bei -80°C aufbewahrt werden müssen und nicht länger als ein Jahr dauern dürfen, während feste Abschnitte viel länger bei Raumtemperatur gelagert werden können. Dies sind einige der wichtigsten Überlegungen, um zu bestimmen, ob formalin-feste Abschnitte von Paraffin-eingebettetem Gewebe oder frische Abschnitte von gefrorenem Gewebe verwendet werden sollen. Letztlich, wenn man genug Gewebe hat, kann es am besten sein, nur einige von beiden zu haben.
In diesem Experiment machten wir uns daran, festzustellen, ob die Cyclin-D1-Expression in vergrößerten Milz aus einem spontanen Mausmodell der Lymphomentwicklung erhöht wurde. Splenic-Gewebeproben wurden zuerst entweder von Wildtyp-Mäusen, transgenen Mäusen ohne Lymphom oder transgenen Mäusen, die spontan Lymphom entwickelt haben, isoliert. Die Milzgewebeproben wurden in Paraformaldehyd fixiert, in Paraffin eingebettet, geschnitten, mit einem Maus-Anti-Cyclin-D1-Primärantikörper, gefolgt von einem Pferde-Anti-Maus-Sekundärantikörper, gebeizt und mit 3,3-Diaminobenzidin (DAB) entwickelt. Die Abschnitte wurden dann in Harris Hematoxylin Solution gegenstainiert und dann wurden die Abschnitte bei 20-facher Vergrößerung abgebildet.
Reagenzien
Paraffin-Eingebettete Abschnitte
- 4% Paraformaldehyd (PFA)
- Ethanol (wasserfrei denaturiert, histologische Grade 100%, 95%, 80%, 75% und 50%). Verdünnung aus 100% Lagerbestand mit doppelt destilliertem Wasser (ddH2O)
- Xylol
- IHC-kompatible Glasrutsche, um sicherzustellen, dass der Gewebeabschnitt während des gesamten Verfahrens befestigt bleibt. IHC-kompatible Glasschieber haben eine spezielle Beschichtung und sind bei mehreren Händlern erhältlich. Wenn Sie ICC ausführen, verwenden Sie eine kammergekammerte Folie. Kammerrutschen ermöglichen es, Zellen in den Kammern auszusäen und in den Inkubator zu legen, bis zellennahe an der Rutsche befestigt sind und die richtige Konfluenz erreichen, an der die Kammern entfernt werden können und die Färbung auf die gleiche Weise wie IHC verlaufen kann.
- paraffin
- 0,3% Wasserstoffperoxid (H2O2)/Methanol: Zur Zubereitung 1 ml 30% H2O2 bis 99 ml Methanol hinzufügen. Bei -20°C lagern
- Antigen-Abrufpuffer: IHC-Citratpuffer pH 6,0
Frische gefrorene Abschnitte
- Optimale Schnitttemperatur (OCT) Einbettverbindung
- Optimale Fixierung: 4% PFA oder Aceton, das auf -20°C abgekühlt wurde
Färbung
- Sperrpuffer: Sollte vom Benutzer bestimmt werden. Ein Beispiel ist Pferdeserum in 1X PBS verdünnt
- Verdünnter Primärantikörper: siehe Herstellerspezifikationen
- Verdünnter biotinylatierter Sekundärantikörper: siehe Herstellerspezifikationen
- Verdünnte Avidin-Horseradish Peroxidase (HRP): Nur zur Peroxidase-Visualisierung. Siehe Herstellerspezifikationen.
- DAB oder ein anderes kompatibles Substrat
- Counterstain (optional)
- Ethanol (wasserfrei denaturiert, histologische Qualität 100% und 95%)
- Xylol
- Organo/Limonene Mount
IHC and ICC have a vast range of applications. For example, one use of IHC is to examine the expression of oncogenes in spontaneous mouse models of tumor development. In Figure 2, we set out to determine if cyclin D1 expression was increased in enlarged spleens in a spontaneous mouse model of lymphoma development. Splenic tissue samples were fixed in paraformaldehyde, embedded in paraffin, sectioned, stained using an anti-cyclin D1 antibody (diluted 1:200 in blocking buffer), and then the sections were imaged at 20X magnification. Cyclin D1 expressing cells are indicated by the reddish-brown color against the blue tissue background. These results suggest that cyclin D1 expression was increased in enlarged spleens, indicating a correlation between cancer development and cyclin D1 expression in this model.
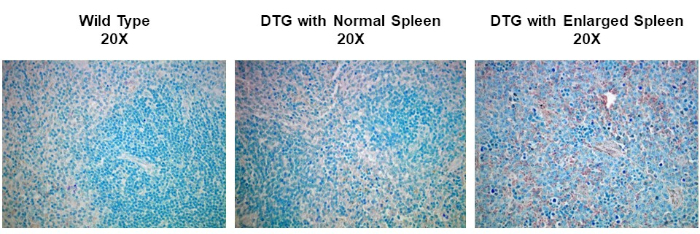
Figure 2: Splenic Cyclin D1 Expression in a Spontaneous Double Transgenic (DTG) Mouse Model of Lymphoma. An image of splenic tissue stained with an anti-Cyclin D1 primary antibody, counterstained with methyl green, and visualized using a biotinylated secondary antibody and ABC reagent activated with DAB substrate. The reddish-brown color represents locations where the antibody has bound indicating the presence of Cyclin D1 expressing tumor cells within the structure of splenic tissue that has been counterstained blue. Please click here to view a larger version of this figure.