Fonte: Michael S. Lee1 e Tonya J. Webb1
1 Departamento de Microbiologia e Imunologia, Faculdade de Medicina da Universidade de Maryland e o Centro de Câncer Integral Marlene e Stewart Greenebaum, Baltimore, Maryland 21201
Imunohistoquímica (IHC) e imunocytoquímica (ICC) são técnicas utilizadas para visualizar a expressão e localização de antígenos específicos usando anticorpos. O primeiro uso publicado do IHC foi em 1941, quando Albert Coons usou a técnica para visualizar a presença de antígeno pneumocócico em seções de tecidos de camundongos infectados com Pneumococcus (1). O nome, imunohistoquímica, é derivado das raízes “imuno-“, em referência a anticorpos, e “histo-“, em referência às seções teciduais usadas no IHC. A raiz “cito-” na imunocitoquímica destaca a diferença fundamental entre ICC e IHC. Enquanto o IHC usa seções de tecido inteiro, o ICC usa células isoladas do tecido ou cultivadas na cultura. A diferença nas amostras utilizadas significa que a preparação da amostra difere tecnicamente entre iHC e ICC, mas caso contrário, os protocolos para ICC e IHC são idênticos e descobrirão que os termos são frequentemente usados de forma intercambiável.
Tanto no IHC quanto no ICC, anticorpos com etiquetas químicas ou fluorescentes, como peroxidase ou rhodamina, respectivamente, são usados para visualizar a distribuição de qualquer antígeno de interesse através de ligação específica do anticorpo marcado ao antígeno. No caso do IHC, finas fatias de tecido são imobilizadas em um slide para manter a estrutura do tecido antes de serem manchadas, permitindo a visualização de antígenos no contexto de tecidos inteiros (Figura 1). No caso do ICC, as células são distribuídas uniformemente em um slide antes de serem manchadas, permitindo a visualização da distribuição de antígenos dentro de células individuais, mas não dentro da estrutura de qualquer tecido específico. Devido às semelhanças entre os dois protocolos, este protocolo se concentrará no IHC para abordar as complexidades adicionais da preparação da amostra envolvida no IHC.
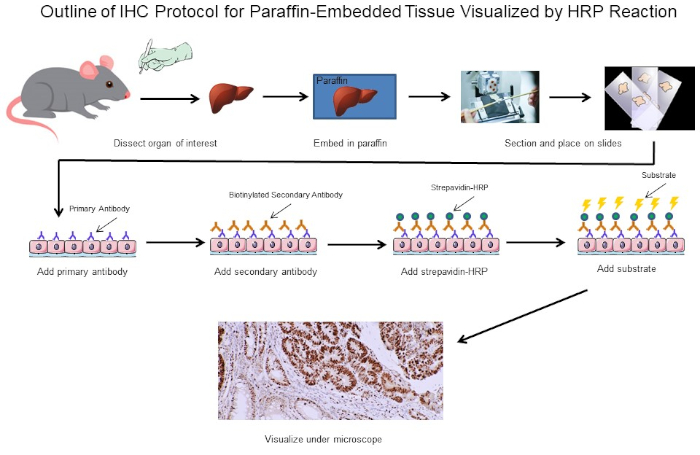
Figura 1: Esboço do Protocolo IHC. Esboço visual de um protocolo IHC para tecido embutido em parafina dissecado a partir de um rato. Este protocolo usa um anticorpo secundário biotinína e strepavidin-HRP para visualizar a localização da ligação de anticorpos. Outras opções, como anticorpos fluorescentes marcados, também são possíveis. Clique aqui para ver uma versão maior desta figura.
A primeira grande decisão ao realizar o IHC é como preparar as seções teciduais para manter a estrutura do tecido durante todo o processo de coloração. As duas principais opções são seções fixadas de formalina de tecido embutido em parafina ou seções frescas de tecido congelado. Não há uma resposta simples sobre qual método usar, pois depende de qual análise a jusante será conduzida. A fixação de formalina de tecidos incorporados para a parafina é geralmente pensada para preservar melhor a morfologia tecidual para uma imagem ideal, enquanto o congelamento de tecido fresco pode preservar a função proteica para ensaios subsequentes fora do IHC. Além disso, seções de tecido congelado fresco têm se mostrado mais adequadas para a análise da expressão genética (2). Uma terceira consideração é se os anticorpos para o seu antígeno de interesse são adequados para seções de tecido fixo ou congelado, pois alguns anticorpos só foram otimizados para um tipo específico de seção e podem não funcionar para outros. Por fim, também é preciso determinar quanto tempo eles precisam para armazenar as seções de tecido, uma vez que amostras congeladas frescas devem ser mantidas a -80°C e podem não durar mais de um ano enquanto seções fixas podem ser armazenadas por muito mais tempo à temperatura ambiente. Estas são algumas das principais considerações para determinar se usar seções fixas de formalina de tecido embutido parafina ou seções frescas de tecido congelado. Em última análise, se alguém tem tecido suficiente, talvez seja melhor ter alguns dos dois.
Neste experimento, nos propusemos a determinar se a expressão cyclin D1 foi aumentada em baços ampliados a partir de um modelo espontâneo de desenvolvimento de linfoma. Amostras de tecido esplênico foram isoladas pela primeira vez de camundongos do tipo selvagem, camundongos transgênicos que não têm linfoma, ou camundongos transgênicos que desenvolveram linfoma espontaneamente. As amostras de tecido de baço foram fixadas em paraformaldeído, embutidas em parafina, seccionadas, manchadas usando um anticorpo primário anti-cíclin D1 seguido por um anticorpo secundário anti-rato de cavalo, e desenvolvidas usando 3,3 diaminobenzidina (DAB). As seções foram então contra-manchadas na Solução de Hematoxilina Harris e, em seguida, as seções foram imagens em ampliação de 20X.
Reagentes
Seções incorporadas à parafina
- 4% paraformaldeído (PFA)
- Etanol (anidro desnaturado, grau histológico 100%, 95%, 80%, 75% e 50%). Pode ser diluído de estoque 100% usando água dupla destilada (ddH2O)
- Xileno
- Lâmina de vidro compatível com IHC para garantir que a seção tecidual permaneça presa durante todo o procedimento. Slides de vidro compatíveis com IHC têm um revestimento especializado e estão prontamente disponíveis em vários varejistas. Se estiver executando o ICC, use um slide com câmara. Os slides de câmara permitem que as células sejam semeadas nas câmaras e colocadas na incubadora até que as células se conectem ao slide e atinjam a confluência adequada, momento em que as câmaras podem ser removidas e a coloração pode proceder da mesma maneira que o IHC.
- Parafina
- 0,3% peróxido de hidrogênio (H2O2)/metanol: Para preparar, adicione 1 mL 30% H2O2 a 99 mL de metanol. Armazenar a -20°C
- Tampão de recuperação de antígeno: PH 6.0 tampão de citrato IHC
Seções congeladas frescas
- Composto de incorporação da temperatura de corte ideal (OCT)
- Fixação ideal: 4% PFA ou acetona que foi resfriado a -20°C
Mancha
- Tampão de bloqueio: Deve ser determinado pelo usuário. Um exemplo é o soro de cavalo diluído em 1X PBS
- Anticorpo primário diluído: veja as especificações do fabricante
- Anticorpo secundário biotinilado diluído: veja as especificações do fabricante
- Visão diluído peroxidase (HRP): Somente para visualização peroxidase. Veja as especificações do fabricante.
- DAB ou outro substrato compatível
- Contra-mancha (opcional)
- Etanol (anidro desnaturado, grau histológico 100% e 95%)
- Xileno
- Monte Organo/Limonene
IHC and ICC have a vast range of applications. For example, one use of IHC is to examine the expression of oncogenes in spontaneous mouse models of tumor development. In Figure 2, we set out to determine if cyclin D1 expression was increased in enlarged spleens in a spontaneous mouse model of lymphoma development. Splenic tissue samples were fixed in paraformaldehyde, embedded in paraffin, sectioned, stained using an anti-cyclin D1 antibody (diluted 1:200 in blocking buffer), and then the sections were imaged at 20X magnification. Cyclin D1 expressing cells are indicated by the reddish-brown color against the blue tissue background. These results suggest that cyclin D1 expression was increased in enlarged spleens, indicating a correlation between cancer development and cyclin D1 expression in this model.
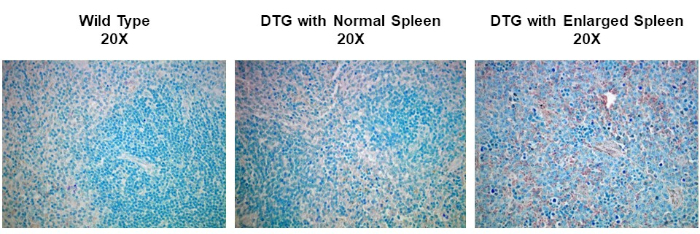
Figure 2: Splenic Cyclin D1 Expression in a Spontaneous Double Transgenic (DTG) Mouse Model of Lymphoma. An image of splenic tissue stained with an anti-Cyclin D1 primary antibody, counterstained with methyl green, and visualized using a biotinylated secondary antibody and ABC reagent activated with DAB substrate. The reddish-brown color represents locations where the antibody has bound indicating the presence of Cyclin D1 expressing tumor cells within the structure of splenic tissue that has been counterstained blue. Please click here to view a larger version of this figure.