Analysis of Neutral Lipid Synthesis in Saccharomyces cerevisiae by Metabolic Labeling and Thin Layer Chromatography
Summary
Here, a protocol is presented for the metabolic labeling of yeast with 14C-acetic acid, which is coupled with thin layer chromatography for the separation of neutral lipids.
Abstract
Neutral lipids (NLs) are a class of hydrophobic, chargeless biomolecules that play key roles in energy and lipid homeostasis. NLs are synthesized de novo from acetyl-CoA and are primarily present in eukaryotes in the form of triglycerides (TGs) and sterol-esters (SEs). The enzymes responsible for the synthesis of NLs are highly conserved from Saccharomyces cerevisiae (yeast) to humans, making yeast a useful model organism to dissect the function and regulation of NL metabolism enzymes. While much is known about how acetyl-CoA is converted into a diverse set of NL species, mechanisms for regulating NL metabolism enzymes, and how mis-regulation can contribute to cellular pathologies, are still being discovered. Numerous methods for the isolation and characterization of NL species have been developed and used over decades of research; however, a quantitative and simple protocol for the comprehensive characterization of major NL species has not been discussed. Here, a simple and adaptable method to quantify the de novo synthesis of major NL species in yeast is presented. We apply 14C-acetic acid metabolic labeling coupled with thin layer chromatography to separate and quantify a diverse range of physiologically important NLs. Additionally, this method can be easily applied to study in vivo reaction rates of NL enzymes or degradation of NL species over time.
Introduction
Acetyl-CoA is the fundamental building block of diverse biomolecules including neutral lipids (NLs), which serve as a versatile biomolecular currency for building membranes, generating ATP, and regulating cell signaling1,2. The availability of NLs to be shunted into any of these respective pathways is, in part, regulated by their storage. Lipid droplets (LDs), cytoplasmic organelles composed of hydrophobic cores of triglycerides (TGs) and sterol-esters (SEs), are the main storage compartments of most cellular NLs. As such, LDs sequester and regulate NLs, which can be degraded and subsequently utilized for biochemical and metabolic processes3,4. It is known that the mis-regulation of NL and LD-associated proteins is correlated with the onset of pathologies including lipodystrophy and metabolic syndromes5,6. Because of this, current LD research is intensely focused on how NL synthesis is regulated spatially, temporally, and across distinct tissues of multi-cellular organisms. Due to the ubiquitous cellular roles for NLs, many enzymes responsible for the synthesis and regulation of NLs are conserved throughout eukaryotes7. Indeed, even some prokaryotes store NLs in LDs8. Therefore, genetically tractable model organisms such as Saccharomyces cerevisiae (budding yeast) have been useful for the study of NL synthesis and regulation.
The separation and quantification of NLs from cell extracts can be accomplished in a myriad of ways, including gas chromatography-mass spectrometry (GC-MS), high-performance liquid chromatography (HPLC), and ultra-performance liquid chromatography-mass spectrometry (UPLC-MS)9,10,11. Perhaps the simplest method for separating NLs is via thin layer chromatography (TLC), which allows for subsequent densitometric quantification from a standard curve12,13. Although TLC provides only a course-grained separation of NLs, it remains a powerful technique because it is inexpensive, and it allows for the rapid separation of NLs from several samples simultaneously. Two of the most considerable challenges facing the study of NLs via TLC are: 1) the broad range of cellular abundances of NL species and their intermediates, and 2) the range of hydrophilicity/hydrophobicity of lipid intermediates within NL synthesis pathways. Consequently, the quantification of NL species via TLC is typically restricted to the most abundant species; however, introduction of a 14C-acetic acid radiolabel can significantly enhance the detection of low abundance intermediates within NL pathways. Acetic acid is rapidly converted into acetyl-CoA by the acetyl-CoA synthetase ACS214, which makes 14C-acetic acid a suitable radiolabeling substrate in yeast15. Additionally, separation of both hydrophobic NLs and hydrophilic intermediates of NLs can be achieved by TLC through the use of multiple solvent systems16. Here, a method is presented for the separation of NLs using 14C-acetic acid metabolic labeling in yeast. Lipids labeled during the pulse period are subsequently isolated by a well-established total lipid isolation protocol17, followed by the separation of NL species by TLC. Developing of TLC plates by both autoradiography to visualize labeled lipids, and a chemical spray to visualize total lipids, permits for multiple methods of quantification. Individual lipid bands can also be easily extracted from the TLC plate using a razor blade, and scintillation counting can be used to quantify amount of radiolabeled material within the band.
Protocol
1. Growth and labeling of yeast cells with 14C-acetic acid
- Inoculate a yeast culture by picking a colony from a plate and dispensing it into 20 mL of synthetic complete (SC) media containing 2% dextrose (see Supplementary File for the recipe of SC media). Incubate at 30 ˚C for overnight with shaking at 200 rpm.
NOTE: Growth condition, sample volume, and treatment will differ based on the lipid(s) of interest. Prior to running full experiments, optimal growth conditions and culture volumes should be empirically determined. This protocol discusses radiolabeling of yeast cultures grown to stationary phase, a growth phase when bio-membrane and cell growth slows, and NL synthesis is very active. - Measure the OD600 of the overnight culture using a spectrophotometer and dilute the yeast cell culture to a final OD600 of 0.2 in 50 mL of fresh SC media containing 2% dextrose. Grow the cells for 24 h, or until they have reached the stationary phase (which is commonly defined by a flat lining of the cell doubling OD600 measurement).
- Before collecting the cells, make the quenching buffer (see Supplementary File for details). Make two 20 mL aliquots of quenching buffer for each sample (i.e., 40 mL quenching buffer for each sample) and split evenly into two 50 mL conical tubes). Store the quenching buffer aliquots at -80 °C for future use.
- Once the cultures have reached the desired OD or growth phase, collect the cells by centrifuging at 4,100 x g for 10 min. While samples are in the centrifuge, prepare radiolabeling media by adding [1-14C] acetic acid sodium salt to dextrose-free SC media at a final concentration of 10 μCi/mL.
CAUTION: Proper personal protective equipment (PPE) should be worn all times when working with radioactive materials. Always follow local guidelines for the proper storage, usage, and disposal of radioactive materials.
NOTE: Both the concentration of 14C-acetic acid in the labeling media and the radiolabeling incubation time should be adjusted according to the metabolite(s) of interest. Here, a 20 min radiolabeling pulse incubation is used, which is sufficient to label NL species with a range of abundance. - Remove the supernatant from the pelleted cells, and wash the cell pellet once with 20 mL of dextrose-free SC media by resuspending the pellet with a pipette. Collect the cells again by centrifuging at 4,100 x g for 5 min.
- Resuspend the cells in 1 mL of dextrose-free SC media, and transfer the cells to a labeled 2 mL microcentrifuge tube. Collect the cells again by centrifuging at 4,100 x g for 2 min.
- Resuspend the cells once more in 500 μL of dextrose-free SC media. Pre-cool a centrifuge equipped for 50 mL conical tubes to -10 °C or on the lowest temperature setting.
- Begin the radiolabeling period by quickly adding 500 μL of radiolabeling media to each 500 μL of cell suspension (final 14C-acetic acid concentration = 5 μCi/mL). Incubate the tubes in a rotating incubator at 30 °C for 20 min. 2 min before the end of the labeling period, transfer one 20 mL aliquot of quenching buffer for each sample from the -80 °C freezer to a bucket of ice
- Once the radiolabeling period has ended, use a pipette to plunge the entire 1 mL sample into 20 mL of cold quenching buffer. Vortex the conical tubes for 5-10 s to ensure that the sample has been thoroughly mixed with the quenching buffer. Incubate the samples in quenching buffer for 2 min on ice.
- Collect the cell pellet by spinning in a centrifuge at 5,000 x g for 3 min set to -10 °C or on the lowest temperature setting. While sample tubes are spinning, transfer another set of quenching buffer aliquot from the -80 °C freezer to a bucket filled with ice (i.e., one 20 mL tube of quenching buffer per sample).
- Remove the quenching buffer supernatant from cell pellets and replace it with 20 mL fresh, cold, quenching buffer. Vortex and shake the samples until the pellet has been dislodged from the bottom of the conical tube and resuspended fully in quenching buffer. Centrifuge the samples again at 5,000 x g for 3 min at -10 °C to collect the cells.
- Once the cells are pelleted, thoroughly remove all quenching buffer from the samples by pouring off the supernatant and removing the excess with a pipette. Store tubes at -80 °C for further processing.
2. Isolation of total lipids from yeast
NOTE: The following protocol for lipid isolation is based on a well-established and frequently used method that efficiently extracts most neutral lipid species17,18.
CAUTION: When using organic solvent, always wear appropriate PPE and work inside of a fume hood when possible. During lipid extraction, avoid using plastics that are incompatible with organic solvents. Polypropylene tubes are suitable for the following protocol.
- Weigh 0.3 g of acid-washed glass beads for each sample and store them in 2 mL microcentrifuge tubes on ice. Remove the cell pellets from the -80 °C freezer and keep them on ice. Add 350 μL methanol and 700 μL chloroform to each sample, resuspend, and transfer to microcentrifuge tubes containing pre-weighed glass beads.
- Lyse cells by agitating tubes on a vortex 3x for 1 min, with 30 s incubations on ice between agitations. Alternatively, cells can be lysed using a mini bead-beater for three 1-min cycles. Save 25-30 μL of whole cell lysate in a separate tube for scintillation counting.
NOTE: The saved lysate will be used to determine the relative amount of radioisotope taken up by each sample during the pulse period, which will influence the amount of each sample loaded onto the TLC plate. This is discussed further in step 3.2. - Pour the entire contents of the 2 mL microcentrifuge tube into a 15 mL glass centrifuge tube [Tube A]. Wash the 2 mL microcentrifuge tubes by adding 1 mL of methanol and vortexing for 10-15 s. Transfer the 1 mL methanol wash to tube A and add 2 mL of chloroform to tube A followed by 400 μL of water for a final sample volume of 4.45 mL.
- Vortex samples for 1 min followed by a 5 min centrifugation at 1,000 x g. After centrifugation, the aqueous (upper) and organic (lower) phases should be clearly visually separated with cell debris lying at the interface.
- Using a glass Pasteur pipette, collect the organic phase from tube A and move to a new 15 mL glass centrifuge tube (Tube B). Add 1 mL of 1M KCl to tube B. To tube A, add 1 mL of methanol and 2 mL of chloroform, and 200 μL MiliQ water. Repeat the vortexing and centrifugation steps on tube A.
- Once again collect the organic phase from tube A and add it to tube B. Dispose of tube A in an appropriate container. Vortex tube B for 1 min followed by a 5 min centrifugation at 1,000 x g.
- Remove the upper aqueous layer from tube B and dispose. Add 1 mL of fresh 1 M KCl back to tube B and repeat the vortexing/centrifugation step. Once layers are separated, carefully collect the entire bottom organic layer into a labeled 4 mL glass vial.
NOTE: At this step, lipid extracts can be stored at -80 °C, or the protocol can be continued for TLC separation of lipids.
3. Separation and quantification of radioisotope-labeled NLs by thin layer chromatography
- If lipid extracts were placed at -80 °C, slowly bring to room temperature by incubating on ice and subsequently on a benchtop. Completely evaporate solvent from lipid extracts by vacuum drying or using a gentle stream of inert gas (e.g., argon or nitrogen). Meanwhile, pre-heat an oven to 145 °C for heating the TLC plate.
- Before samples can be loaded onto the TLC plate, determine relative amounts of radiolabel taken up by the cells. Pipette 10 μL of the whole cell lysate from step 2.2 into a 6 mL glass scintillation vial, add 6 mL of scintillation fluid and place vials in a rack. Use a scintillation counter to measure the cpm or dpm of each sample using the count single rack option set to a 1 min counting time. Measure each whole cell lysate in duplicate to obtain an average for each sample. Adjust the loading amount according to a wild type or reference sample
NOTE: The amount of each sample to load onto the TLC plate can be determined using the following equation: (average sample counts)/(average reference counts) x desired loading volume. For example, if 20 μL of the reference sample is to be loaded onto the TLC plate, and has an average count of 1,000, then an experimental sample with an average count of 2,000 will have 10 μL loaded onto the TLC plate. - Reconstitute the sample lipids in 40-50 μL of 1:1 (v/v%) chloroform:methanol by vortexing for 5 min. Prepare 101 mL of the mobile phase solvent in a glass graduated cylinder (see the Representative Results section for an example of major NL species separation by a 50:40:10:1 (v/v/v/v%) Hexane:Petroleum ether:Diethyl ether:Acetic acid solvent).
- Pour the solvent into a glass TLC chamber containing a 20 x 20 TLC saturation pad and a tight-fitting lid. Prepare a channeled 20 x 20 silica gel 60 G plate by gently marking a line 1.5 cm above the bottom of the plate using a pencil. The line designates the origin and where the lipids will be loaded. Below the line, gently label the sample that will be loaded in each lane. Once the TLC plate has been prepped, incubate the plate in a 145 °C oven for at least 30 min to pre-heat the plate and remove any excess moisture.
- Once the plate has been sufficiently heated, and the TLC saturation pad is saturated with solvent, remove the TLC plate from the oven and immediately proceed to loading the TLC plate. Loading the plate while it is warm ensures rapid solvent evaporation. For each lipid species of interest, load 5-20 μg of a purified lipid standard onto a lane of the TLC plate to track separation and expected migration distance. Using a pipette, spot 5μL of sample onto the origin of each lane located 1.5 cm above the bottom the of TLC plate. Repeat loading of 5 μL spots until 20-40 μL of sample has been loaded into each lane.
NOTE: 5-20 μg of unlabeled purified lipids can be added to each sample lane as tracers that can be stained and visualized following TLC separation of lipids. The presence of a stained standard allows for easy tracking and excision of radiolabeled lipid bands for subsequent scintillation counting. Which purified standards are loaded onto the plate will be determined by the NL species of interest. See the Representative Result section for examples of separating oleic acid (FFA), 1,2 dioleoyl-glycerol (DG), triolein (TG), cholesterol (Chol), cholesteryl-linoleate (SE), and squalene in lanes adjacent to the sample lanes. - Once the standard and the experimental samples have been loaded, place the plate in the developing chamber and wait until the solvent has reached the top of the plate (40-60 min). Once the plate is fully developed, remove it from the chamber and allow it to dry in the fume hood for 20 minutes.
- After the plate is dried, cover it with plastic film and place it in a developing cassette with an autoradiography screen. Allow the plate to develop with the screen for 24-48 h.
4. Visualization and quantification of TLC separated lipids
- Remove the screen from the developing cassette and place inside of a phosphor imager. Select the Phosphor Imaging option and develop at 800-1000 V.
NOTE: Phosphor imaging gives a qualitative view of radiolabeled lipids on the TLC plate. However, quantification of radiolabeled lipids is best accomplished by scintillation counting, which is described subsequently. - Mix 100 mL of p-anisaldehyde reagent (see Supplementary File) and deposit in a glass spray bottle. Spray the TLC plate with p-anisaldehyde reagent until the silica is saturated. Bake the plate in a 145 °C oven for 5 min, or until bands have appeared.
- To quantify individual lipid species using radiolabel scintillation counting, use a razor blade to scrape the silica gel from the glass TLC plate. Transfer each silica gel band corresponding to a single radiolabeled lipid species to a glass scintillation vial and add 6 mL scintillation fluid. Vortex vigorously until the silica band has been reduced to small pieces.
- Alternatively, lipids can be extracted from the silica gel band using the lipid extraction protocol in section 3. If lipids are extracted from the silica gel, evaporate the solvent entirely as in step 3.1, and add 6 mL scintillation fluid to the dried lipids. Place the rack containing scintillation vials into a scintillation counter. Select the Count single rack option and adjust the counting time to 2 min per vial. Results from the scintillation counter will be printed and can be visualized as a bar graph.
Representative Results
In this protocol, we have demonstrated that the labeling, detection, and quantification of NL species can be accomplished by 14C-acetic acid metabolic labeling. Major NL species can be separated in a solvent system of 50:40:10:1 (v/v/v/v%) Hexane:Petroleum ether:Diethyl ether:Acetic acid (Figure 1A,B). Phosphor imaging allows for visualization of labeled free fatty acid (FFA), triacylglycerol (TG), diacylglycerol (DG), cholesterol (Chol), and squalene (SQ) (Figure 1A). Although SEs can be separated from other NL species in this solvent, none are detected in the autoradiogram following a 20-minute pulse. This may be attributed to slow SE synthesis during the stationary phase of growth in yeast. It is also demonstrated that purified lipid species can be separated in this method and subsequently visualized by spraying of the TLC plate with p-anisaldehyde reagent (Figure 1B). While NL species are well separated in this solvent, polar species like phosphatidylcholine (PC) stay at the origin (Figure 1B). By applying a chase period in radiolabel-free media following the pulse, relative flux through NL pathways can be measured (Figure 1C). After a 10-min chase, the major pool of SQ has disappeared, and total Chol is elevated. Similarly, the appearance of DG in the chase period correlates with a decrease in the FFA signal.
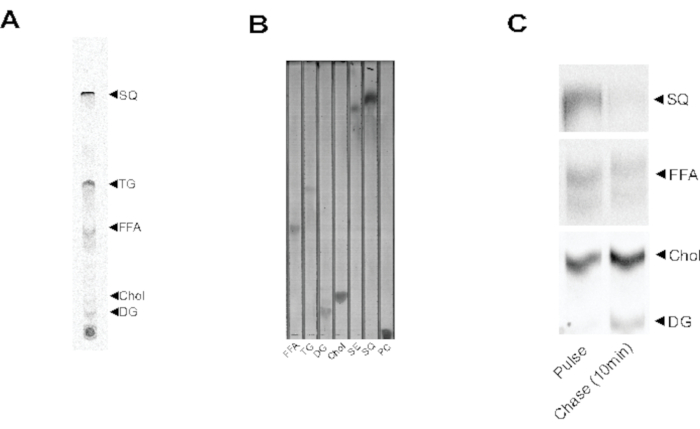
Figure 1: 14C-Acetic acid radiolabeling allows for detection of multiple NL species. (A) Autoradiogram of lipids separated by TLC isolated from yeast radiolabeled with 14C-acetic acid in stationary phase. Clearly detectable species include free fatty acid (FFA), triglyceride (TG), diacylglycerol (DG), cholesterol (Chol), and squalene (SQ). Unlabeled bands are unidentified NL species. (B) Purified lipid species separated by TLC and visualized by p-anisaldehyde staining. Visualized species include all lipids mentioned in (A) in addition to sterol-esters (SE) and phosphatidylcholine (PC). (C) Autoradiogram of lipids separated by TLC isolated from yeast pulsed with 14-acetic acid in stationary phase followed by a 10 min chase period in radiolabel-free media. Disappearance of SQ is met with increase in Chol. Rise in DG in the chase period is accompanied by decrease in FFA species. Please click here to view a larger version of this figure.
Supplementary File: Recipes for buffers, media, and solutions. Please click here to download this file.
Discussion
Here, a versatile radiolabeling protocol to quantitatively monitor the synthesis of NL species in yeast is presented. This protocol is very modular, which allows for the procedure to be finished within 3-6 days. Additionally, a wealth of literature exists on the use of TLC to separate lipid species and metabolites, which should permit the user to detect several lipid species of interest with a simple change of TLC solvent systems16,19.This protocol is conducive to the separation, detection, and quantification of radiolabeled lipids. It can also be coupled with a chase period in un-labeled media to detect the turnover time of labeled NLs. Collectively, this procedure gives a useful structure to begin exploring the radiolabeling of NL species.
Other methods, such as HPLC, GC-MS, and UPLC-MS provide higher resolution of lipid separation and quantification; however, it is typically not optimal to run radiolabeled samples through MS, although this can be overcome by using stable-isotopes. Nevertheless, this radiolabel method provides high detection sensitivity and versatility for many lipid species. Another advantage of this protocol compared to MS is its affordability. TLC separation of lipids is relatively simple, requires no extravagant equipment, and relies on common laboratory materials. Regarding limitations: certain low-abundance species, like lyso-lipids, may not be detectable even following incorporation of a 14C label. Additionally, most TLC approaches are not suitable for 'lipidomic' characterizations, due to the course-grained separation of lipid species within a given solvent.
Yeast offer a convenient, genetically tractable model system for the study of lipids via radiolabel biochemical approaches. However, it should be noted that in specific genetic backgrounds, or during a particular metabolic growth conditions, the cellular uptake of radiolabeled-acetic acid or other radiolabels may be reduced. Labeling of cells with 14C-acetic acid in the absence of glucose robustly increases the uptake of the radiolabel. Long incubations in the absence of glucose will proportionally increase radiolabel uptake; however, this may also influence the pathways in question. Therefore, labeling efficiency for a particular growth condition should be established prior to following the 14C-acetic acid radiolabeling protocol in full. In particular, pay attention to the length of the radiolabeling period. The labeling time should be kept as short as possible to detect the lipid species-of-interest. Altogether, this procedure allows for the study of important lipid synthesis reactions and should permit the investigation of NL regulation in intact cells.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors would like to thank the members of the Henne lab for help and conceptual advice in the completion of this study. W.M.H. is supported by funds from the Welch Foundation (I-1873), the NIH NIGMS (GM119768), the Ara Paresghian Medical Research Fund, and the UT Southwestern Endowed Scholars Program. S.R has been supported by a T32 program grant (5T32GM008297).
Materials
| [1-C14] Acetic acid sodium salt specific activity: 45-60mCi | PerkinElmer | NEC084H001MC | |
| 18:1 1,2 dioleoyl-sn-glycerol | Avanti | 800811O | |
| 200 proof absolute ethanol | Sigma | 459836 | |
| Acid washed glass beads 425-600um | Sigma | G8772 | |
| Amber bulbs for Pastuer pipettes | Fisher | 03-448-24 | |
| Ammonium Sulfate >99% | Sigma | A4418 | |
| Beckman LS6500 scintillation counter | PerkinElmer | A481000 | |
| Chloroform (HPLC grade) | Fisher | C607SK | |
| Cholesterol >99% | Sigma | C8667 | |
| Cholesteryl-linoleate >98% | Sigma | C0289 | |
| Concentrated sulfuric acid | Sigma | 339741 | |
| Corning 50mL conical tubes, polypropylene with centristar cap | Sigma | CLS430829 | |
| Dextrose, anhydrous grade | Sigma | D9434 | |
| Diethyl ether anhydrous grade | Sigma | 296082 | |
| Drying oven | Fisher | 11-475-155 | |
| EcoLume scintillation liquid | VWR | IC88247001 | |
| Eppendorf 5424R centrifuge | Fisher | 05-401-205 | |
| GE Storage phosphor screen | Sigma | GE28-9564-75 | |
| GE Typhoon FLA9500 imager | |||
| Glacial acetic acid, ACS grade | Sigma | 695092 | |
| Glass 6mL scintillation vials | Sigma | M1901 | |
| Glass centrifuge tube caps | Fisher | 14-595-36A | |
| Glass centrifuge tubes | Fisher | 14-595-35A | |
| Glass Pasteur pipette | Fisher | 13-678-20C | |
| Hexane, anhydrous grade | Sigma | 296090 | |
| L-Adenine >99% | Sigma | A8626 | |
| L-Alanine >98% | Sigma | A7627 | |
| L-Arginine >99% | Sigma | A1270000 | |
| L-Asparagine >98% | Sigma | A0884 | |
| L-Aspartate >98% | Sigma | A9256 | |
| L-Cysteine >97% | Sigma | W326305 | |
| L-Glutamic acid monosodium salt monohydrate >98% | Sigma | 49621 | |
| L-Glutamine >99% | Sigma | G3126 | |
| L-Glycine >99% | Sigma | G8898 | |
| L-Histidine >99% | Sigma | H8000 | |
| L-Isoleucine >98% | Sigma | I2752 | |
| L-Leucine >98% | Sigma | L8000 | |
| L-Lysine >98% | Sigma | L5501 | |
| L-Methionine, HPLC grade | Sigma | M9625 | |
| L-Phenylalanine, reagent grade | Sigma | P2126 | |
| L-Proline >99% | Sigma | P0380 | |
| L-Serine >99% | Sigma | S4500 | |
| L-Theronine, reagent grade | Sigma | T8625 | |
| L-Tryptophan >98% | Sigma | T0254 | |
| L-Tyrosine >98% | Sigma | T3754 | |
| L-Uracil >99% | Sigma | U0750 | |
| L-Valine >98% | Sigma | V0500 | |
| Methanol, ACS grade | Fisher | A412 | |
| Oleic acid >99% | Sigma | O1008 | |
| p-anisaldehyde | Sigma | A88107 | |
| Petroleum ether, ACS grade | Sigma | 184519 | |
| Phosphatidylcholine, dipalmitoyl >99% | Sigma | P1652 | |
| Pipettes | Eppendorf | 2231000713 | |
| Potassium chloride, ACS grade | Sigma | P3911 | |
| Sodium Hydroxide pellets, certified ACS | Fisher | S318-100 | |
| Squalene >98% | Sigma | S3626 | |
| Succinic Acid crystalline/certified | Fisher | 110-15-6 | |
| TLC saturation pad | Sigma | Z265225 | |
| TLC silica gel 60G glass channeled plate | Fisher | NC9825743 | No fluorescent indicators |
| Transparency plastic film | Apollo | 829903 | |
| Tricine | Sigma | T0377 | |
| Triolein >99% | Sigma | T7140 | |
| Vortex mixer | Fisher | 02-215-414 | |
| Whatman exposure cassette | Sigma | WHA29175523 | |
| Yeast nitrogen base without ammonium sulfate and amino acids | Sigma | Y1251 |
References
- Konige, M., Wang, H., Sztalryd, C. Role of adipose specific lipid droplet proteins in maintaining whole body energy homeostasis. Biochimica Et Biophysica Acta. 1842 (3), 393-401 (2014).
- Arrese, E. L., Saudale, F. Z., Soulages, J. L. Lipid droplets as signaling platforms linking metabolic and cellular functions. Lipid Insights. 7, 7-16 (2014).
- Walther, T. C., Chung, J., Farese, R. V. Lipid droplet biogenesis. Annual Review of Cell and Developmental Biology. 33, 491-510 (2017).
- Olzmann, J. A., Carvalho, P. Dynamics and functions of lipid droplets. Nature Reviews. Molecular Cell Biology. 20 (3), 137-155 (2019).
- Krahmer, N., Farese, R. V., Walther, T. C. Balancing the fat: lipid droplets and human disease. EMBO Molecular Medicine. 5 (7), 973-983 (2013).
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature. 362 (6423), 801-809 (1993).
- Zhang, C., Liu, P. The lipid droplet: A conserved cellular organelle. Protein & Cell. 8 (11), 796-800 (2017).
- Wältermann, M., et al. Mechanism of lipid-body formation in prokaryotes: How bacteria fatten up: Lipid-body formation in prokaryotes. Molecular Microbiology. 55 (3), 750-763 (2004).
- Borrull, A., López-Martínez, G., Poblet, M., Cordero-Otero, R., Rozès, N. A simple method for the separation and quantification of neutral lipid species using GC-MS. European Journal of Lipid Science and Technology. 117 (3), 274-280 (2015).
- Kotapati, H. K., Bates, P. D. Normal phase HPLC method for combined separation of both polar and neutral lipid classes with application to lipid metabolic flux. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences. 1145, 122099 (2020).
- Knittelfelder, O. L., Weberhofer, B. P., Eichmann, T. O., Kohlwein, S. D., Rechberger, G. N. A versatile ultra-high performance LC-MS method for lipid profiling. Journal of Chromatography B. 951-952, 119-128 (2014).
- Ruiz, J. I., Ochoa, B. Quantification in the subnanomolar range of phospholipids and neutral lipids by monodimensional thin-layer chromatography and image analysis. Journal of Lipid Research. 38 (7), 1482-1489 (1997).
- Bui, Q., Sherma, J., Hines, J. K. Using high performance thin layer chromatography-densitometry to study the influence of the prion [RNQ+] and its determinant prion protein Rnq1 on yeast lipid profiles. Separations. 5 (1), 5010006 (2018).
- Pronk, J. T., de Steensma, H., Van Dijken, J. P. Pyruvate metabolism in Saccharomyces cerevisiae. Yeast. 12 (16), 1607-1633 (1996).
- Buttke, T. M., Pyle, A. L. Effects of unsaturated fatty acid deprivation on neutral lipid synthesis in Saccharomyces cerevisiae. Journal of Bacteriology. 152 (2), 747-756 (1982).
- Touchstone, J. C. Thin-layer chromatographic procedures for lipid separation. Journal of Chromatography B: Biomedical Sciences and Applications. 671 (1-2), 169-195 (1995).
- Folch, J., Lees, M., Sloane Stanley, G. H. A simple method for the isolation and purification of total lipides from animal tissues. The Journal of Biological Chemistry. 226 (1), 497-509 (1957).
- Breil, C., Abert Vian, M., Zemb, T., Kunz, W., Chemat, F. “Bligh and Dyer” and Folch methods for solid-liquid-liquid extraction of lipids from microorganisms. Comprehension of solvatation mechanisms and towards substitution with alternative solvents. International Journal of Molecular Sciences. 18 (4), 708 (2017).
- Fuchs, B., Süß, R., Teuber, K., Eibisch, M., Schiller, J. Lipid analysis by thin-layer chromatography-A review of the current state. Journal of Chromatography A. 1218 (19), 2754-2774 (2011).
